Чистое помещение (Clean rooms) – искусственная контролируемая среда, специально спроектированная для минимизации концентрации загрязняющих веществ в воздухе, таких как пыль, переносимые по воздуху микробы и аэрозольные частицы.
Главная задача при эксплуатации функционирующего чистого помещения – поддержание в нем соответствующей чистоты.
Чем чувствительнее к контаминантам производимая продукция или производственный процесс, тем выше требования к уровню чистоты воздушного пространства.
На сегодняшний день без чистых помещений невозможно представить такие отрасли промышленности, как электроника, аэрокосмическая отрасль, производство медицинского и лабораторного оборудования, биотехнологии и, безусловно, фармацевтическая промышленность.
Фармацевтическая промышленность – наиболее чувствительная отрасль с точки зрения ответственности за качество содержания и эксплуатации чистых помещений, ведь на кону стоит здоровье и жизнь человека. Даже незначительные нарушения и отклонения от принятых норм и стандартов, в вопросах обслуживания чистых помещений и условиях работы в них, могут нанести непоправимый вред не только потребителю, но и фармацевтическому производителю.
 КЛАССЫ ЧИСТОТЫ ПОМЕЩЕНИЙ
КЛАССЫ ЧИСТОТЫ ПОМЕЩЕНИЙ
Дифференциация классов чистоты помещений выполняется исходя из количества частиц заданного размера на одну единицу воздуха.
Являясь важнейшей характеристикой чистых помещений, этот параметр регулируется стандартом ISO 14644–1, принятым в РФ как ГОСТ Р ИСО 14644–1–2017 «Чистые помещения и связанные с ними контролируемые среды, часть 1. Классификация чистоты воздуха по концентрации частиц». Настоящий стандарт включает в себя чистые помещения классов ISO 1–9, где класс ISO 1 является самым чистым, а класс ISO 9 – самым грязным классом (тем не менее такое помещение все равно чище обычного помещения).
Кроме того, чистые помещения должны соответствовать установленным отраслевым и региональным стандартам. Например, в фармацевтической промышленности применяется Руководство по надлежащей производственной практике ЕС (А-В-С-D).
ЧЕЛОВЕК – ОСНОВНОЙ ИСТОЧНИК ЗАГРЯЗНЕНИЯ
Несмотря на то, что уровень загрязнения воздуха зависит от происходящих в помещении процессов, из-за которых образуются частицы самым большим источником загрязнения, безусловно, является человек.
Человек не может быть стерильным и даже когда он неподвижен, он продолжает распространять вокруг себя десятки миллионов частиц различного размера (частицы омертвевшей кожи, волос), микроорганизмов и других загрязняющих веществ.
Поскольку полностью устранить человека из процесса фармацевтического производства невозможно, следует понимать, что ключевой способ контроля этого вида загрязнений в чистых помещениях заключается в использовании специальной технологичной одежды, которая будет выполнять роль фильтра.
Если сотрудники фармацевтического предприятия, работающие в зоне чистых помещений, не будут одеты соответствующим образом, то прекрасно сконструированное и оснащенное чистое помещение высокого класса в считанные минуты превратится в обычное бытовое помещение.
 ОДНОРАЗОВАЯ СПЕЦОДЕЖДА
ОДНОРАЗОВАЯ СПЕЦОДЕЖДА
Важно учитывать качество используемой спецодежды, чтобы она могла выполнять свои ключевые функции:
- Защищать технологические процессы, продукты и окружающую среду от загрязнений, выделяемых человеком;
- Оберегать человека от вредного влияния окружающей среды, в т. ч. от технологических процессов, используемых в нем материалов и продуктов производства;
- Создавать и поддерживать комфортные условия для персонала.
Основная доля успеха в выполнении ключевых функций спецодежды для чистых помещений приходится на структуру и качество используемого в изготовлении спецодежды материала.
В производстве спецодежды для чистых помещений используют нетканые материалы из синтетических непрерывных (филаментных) нитей. Чаще других используются полиэфирные ткани. Такие материалы имеют минимальное собственное пыление и высокую абразивную устойчивость, а также удовлетворительные антистатические свойства.
Необходимо обращать внимание на тот факт, что одежда для чистых помещений, будучи фильтром загрязнений, имеет высокие значения теплового сопротивления и препятствует нормальному воздухо- и теплообмену между человеком и окружающей средой. Значит, помимо кондиционирования воздуха в чистом помещении стоит обращать внимание на свойства ткани, особенно на паропроницаемость (сколько лишней энергии из пододёжного пространства может выпустить ткань). Нарушение подобного процесса приводит к дискомфорту, снижению внимания человека, повышенной потливости, что увеличивает уровень загрязнения в чистом помещении и уменьшает качество работы персонала.
Вот почему так важен обдуманный подход к выбору спецодежды для чистых помещений, который должен основываться на качестве материала, эргономичности кроя и подтвержденных физико-механических свойствах изделия.

ОДНОРАЗОВАЯ СПЕЦОДЕЖДА ISOGARM™
В 2021 году компания Cleantech® запустила собственное российское производство высококачественной одноразовой спецодежды для чистых помещений класса ISO 4 (GMP A-B) – Isogarm™, отвечающей всем сов-ременным требованиям, предъявляемым к такому типу спецодежды.
По своему качеству спецодежда Isogarm™ не уступает, а в некоторых случаях даже превосходит мировые бренды одноразовой спецодежды.
КАЧЕСТВО ИСПОЛЬЗУЕМЫХ МАТЕРИАЛОВ
Основную роль в уровне качества любой одноразовой спецодежды для чистых помещений играют свойство и структура используемого материала.
Специалисты компании Cleantech® разработали оптимальный для одноразовой спецодежды материал AirCleanTech™, который надежно защищает как человека, так и производственные процессы, и при этом минимально препятствует нормальному воздухо- и парообмену.
Материал AirCleanTech™, из которого производится спецодежда Isogarm™, – это современный нетканый мембранный материал, который имеет оптимальное соотношение плотности, легкости, прочности, воздухо-и парообмена. Материал AirCleanTech™ прошел все необходимые испытания физико-механических свойств.
Кроме того, материал AirCleanTech™ имеет сверхнизкое выделение частиц, что снижает риски контаминации чистых помещений.
Работать в спецодежде из такого материала не только безопасно, но и комфортно.
ОРГАНИЗАЦИЯ ПРОИЗВОДСТВЕННОГО ПРОЦЕССА
Стоит отметить высокий уровень организации производственного процесса компании Cleantech®.
Чтобы обеспечить наивысшее качество и чистоту изделий Isogarm™, на производстве, с заданной периодичностью, контролируются все критические параметры среды. Компания придерживается строгих стандартов качества на каждом производственном этапе, а выпускаемую продукцию сертифицируют органы государственного контроля.
В производстве спецодежды Isogarm™ используется современное безмаслянное швейное оборудование. Само производство спецодежды организовано в чистых помещениях класса чистоты ISO 7 (GMP C-D), а упаковка происходит в помещениях класса чистоты ISO 4 (GMP A-B). Это позволяет соблюдать сверхнизкое содержание и выделение частиц, а также сверхнизкую бионагрузку перед стерилизацией.
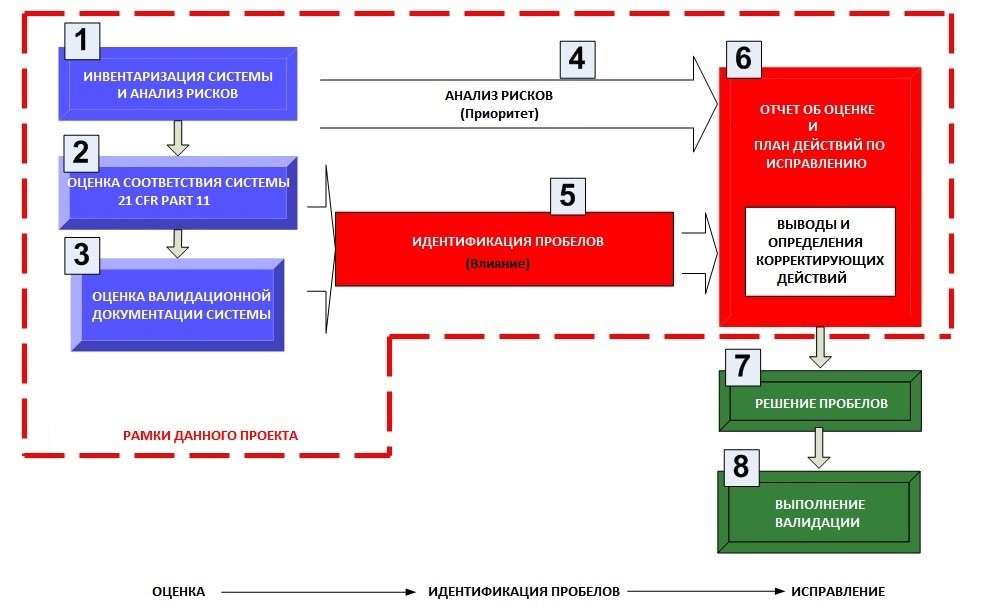
 ВАЛИДАЦИЯ ПРОЦЕССА СТЕРИЛИЗАЦИИ
ВАЛИДАЦИЯ ПРОЦЕССА СТЕРИЛИЗАЦИИ
Стерильные изделия – это изделия свободные от жизнеспособных микроорганизмов. Производство одноразовой спецодежды для чистых помещений происходит в контролируемых условиях с соблюдением всех необходимых правил и требований к чистоте и гигиене, что минимизирует случайную контаминацию продукции, но даже в таких случаях, когда производственный процесс стандартизирован, производимая продукция может содержать определенное (как правило небольшое) количество микроорганизмов перед стерилизацией.
Стандарты и системы управления качеством признают, что эффективность некоторых процессов, применяемых в производстве, не может быть полностью доказана последующими инспекциями и испытанием продукции. Стерилизация продукции является примером такого процесса.
Именно поэтому очень важно, чтобы процесс стерилизации производимой продукции был надлежащим образом валидирован и находился под постоянным контролем. Соответствие данному требованию обеспечивает надежность и воспроизводимость процесса стерилизации, что с достаточной уверенностью обеспечивает минимальный уровень вероятности сохранения микро-организмов в продукции после стерилизации.
Процесс стерилизации спецодежды Isogarm™ валидирован в соответствии с ГОСТ Р ИСО 11137–1–2011. На стерильную продукцию предоставляется протокол, подтверждающий стерильность изделий и гарантирующий уровень обеспечения стерильности 10–6.
КВАЛИФИКАЦИЯ РАБОТАЮЩИХ СПЕЦИАЛИСТОВ
Весь производственный персонал, задействованный в процессе изготовления продукции Isogarm™, имеет высокую квалификацию. Каждый сотрудник производства прошел специальную подготовку и обучение для работы в чистых помещениях и использует средства индивидуальной защиты.
ИНДИВИДУАЛЬНЫЙ ПОДХОД
В силу того, что у каждой фармацевтической компании есть свои особенности и потребности, компания Cleantech® предоставляет индивидуальный подход к каждому клиенту и готова произвести защитную спецодежду с логотипом компании-заказчика или индивидуальными размерами, учитывая морфологические особенности сотрудников клиента.
СВОБОДНАЯ ЛОГИСТИКА
Производство спецодежды Isogarm™ находится в России в городе Санкт-Петербурге, что позволяет производителю оперативно реагировать на потребности клиента. Процесс производства и доставки продукции максимально оптимизирован, что обеспечивает выгодную стоимость и быструю доставку.
СТАНДАРТ КАЧЕСТВА И КОМФОРТА
Одноразовая спецодежда Isogarm™ соответствует всем современным стандартам качества, безопасности, чистоты и стерильности, что очень важно для высокотехнологичных отраслей промышленности, таких как фармацевтическая, биотехнологическая и микроэлектроника.
Использование на производстве одноразовой спецодежды Isogarm™ улучшает качество работы и, как следствие, производительность персонала, поскольку работать в спецодежде Isogarm™ комфортно и безопасно.
Уникальный материал AirCleanTech™, эргономичный крой и каждая продуманная деталь изделий, делают спецодежду Isogarm™ надежным помощником в реализации амбициозных планов любого фармацевтического предприятия. Комфорт и надежность спецодежды Isogarm™ способны увеличить качество и производительность труда персонала.

Контакты:
ООО «КЛИНТЕХ»
Адрес: Санкт-Петербург, Дорога в Каменку, д. 74, Лит. А, помещение 403
Сайт: https://cleanth.ru/
Е-mail: sale@cleanth.ru
Телефон: +7 812 748 25 15









 АО «Асептические медицинские системы»
АО «Асептические медицинские системы»

















 ОДНОРАЗОВАЯ СПЕЦОДЕЖДА
ОДНОРАЗОВАЯ СПЕЦОДЕЖДА ВАЛИДАЦИЯ ПРОЦЕССА СТЕРИЛИЗАЦИИ
ВАЛИДАЦИЯ ПРОЦЕССА СТЕРИЛИЗАЦИИ