

Владимир Орлов, директор Евразийского отделения ISPE
Согласно исследованию ISPE ЕАЭС по цифровизации фармацевтической промышленности, ни один участник тематического опроса не оценил уровень компьютеризации GхP-критичных процессов в своей компании как «полная цифровизация». Бумажный документооборот продолжает господствовать в производственных процессах, при обучении персонала, управлении самоинспекциями и отклонениями, изменениями, при подготовке обзоров по качеству продукции. С чем это связано и какие GxP-требования сегодня предъявляют регуляторы разных стран к компьютеризированным системам в фармкомпаниях?
Регуляторные GxP-требования к компьютеризированным системам (КС) формировались планомерно вслед за внедрением тех или иных IT–инноваций в фармпромышленности. Здесь обращает внимание на себя именно тот факт, что отличительной чертой процесса формирования регулирующих требований к КС является их «запаздывание» относительно фактического внедрения и начала практического использования новых систем с неуклонно расширяющимся функционалом. Процесс формирования нормативного регулирования схематично изображен на Рисунке 1.
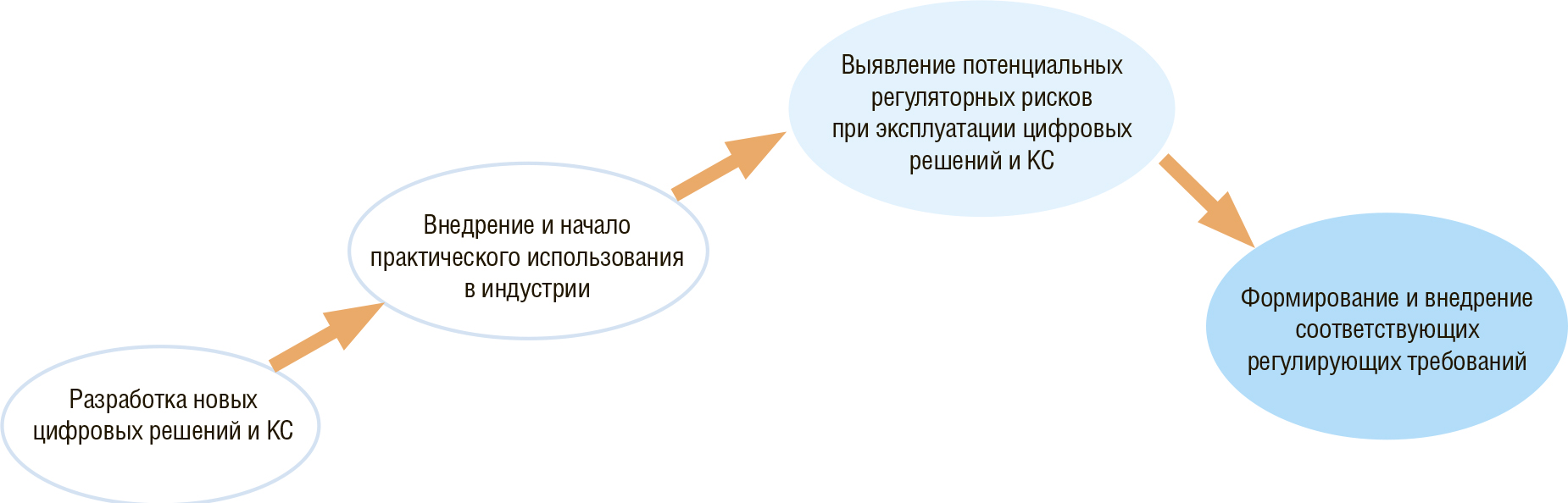
Рис. 1. Процесс формирования нормативного регулирования
Фактически ситуация складывается таким образом, что фармпроизводители, как «конечные» (“end-users”) пользователи КС, не лимитированы в плане использования новаторских решений в IT-среде, которые ещё на момент их внедрения по ряду причин могут быть вне сферы действия существующих регуляторных требований. В таком случае ответственность по реализации требований по обеспечению качества и комплаенс ложится на плечи как разработчиков IT-систем, так и их «конечных» пользователей. Согласно общепринятой практике, в данной ситуации,при отсутствии строго очерченных регуляторных требований и норм, на первое место выходит процесс управления рисками как один из ключевых инструментов фармацевтической системы качества.
Однако, следует отметить, что, несмотря на указанный выше факт некоторого «рассинхрона» между поступательным развитием КС в фармотрасли (как следствие научно-технического прогресса) и формированием регулирующих требований, опыт последних 10–15 лет показывает также совершенствование нормативно-правовой базы в данном направлении. Ниже представлены основные существующие на сегодняшний день регулирующие документы в части использования КС в GxP-регулируемых средах с разбивкой по основным регуляторным системам в мире. Рассматриваемые документы включают в себя как непосредственно требования к самим КС, так и принципы управления целостностью данных (Data Integrity), которые на сегодняшний день неразрывно связаны между собой:
1. Европейский союз:
— ключевые регуляторные требования к используемым КС сформулированы в Приложении № 11 к GMP ЕС, которое так и называется – «Computerised Systems». Текущая версия документа действует с 2011 г. и в настоящий момент находится в процессе пересмотра;
— на использование КС в части хранения записей и данных распространяется также действие Главы № 4 GMP ЕС – «Документация»;
— относительно производства фармацевтических субстанций (АФС) основные требования к КС приведены в подразделе 5.4 Части II GMP ЕС.
2. Схема взаимодействия фармацевтических инспекций – PIC/S:
— требования, указанные в Приложении №11, Главе № 4 и Части II GMP PIC/S, по сути, идентичны GMP ЕС;
— помимо указанного выше, в рамках PIC/S были изданы два руководства – Good Practices for Computerised Systems in Regulated “GxP” environments (2007 год) и Good Practices for Data Management and Integrity in Regulated GMP/GDP Environments. Первый из указанных документов уже, безусловно, устарел (хотя и является формально действующим), а второй, опубликованный в 2021 г., является актуальным и на сегодняшний день.
3. Всемирная Организация Здравоохранения – ВОЗ (WHO):
— В 2021 году ВОЗ в составе Технического отчёта № 1033 опубликовала обновлённое руководство «Guideline on data integrity», которое заменило собой существовавший с 2016 г. документ «Guidance on good data and record management practices». Основной целью пересмотра документа явилась необходимость его гармонизации с другими международными руководствами в данной сфере – PIC/S, MHRA и FDA (рассмотрены ниже).
4. Управление по контролю лекарственных средств и изделий медицинского назначения Соединённого Королевства (MHRA)
— «‘GXP’ Data Integrity Guidance and Definitions» – руководство, опубликованное в 2018 г., в большей степени ориентировано именно на управление целостностью данных, нежели на вопросы использования КС. Однако, как было сказано выше, на сегодняшний день они тесно взаимосвязаны между собой.
5. Управление по контролю за качеством пищевой продукции и лекарственных средств США (US FDA):
— Основные требования, предъявляемые к КС и целостности данных, указаны в соответствующих разделах CFR Title 21 Parts 210, 211, 212 (Свод нормативных актов федеральных органов исполнительной власти США, Глава 21 ч. 210, 211, 212). Однако, следует отметить, что указанная в данных разделах информация является «обобщённой», поэтому в дополнение к ней было разработано отдельное Руководство (ниже);
— В 2018 г. US FDA опубликовало Руководство для фармотрасли «Data Integrity and Compliance With Drug CGMP Questions and Answers Guidance for Industry», в котором сформулированы разъяснения касательно регуляторных требований, указанных в CFR Title 21 Parts 210, 211, 212. Как указано в преамбуле Руководства, его принципы распространяются на процессы производства всех лекарственных средств, включая биологические препараты, а также АФС.
6. Российская Федерация и ЕАЭС:
— на сегодняшний день регуляторные требования к КС и обеспечению целостности данных сформулированы в Приложении № 11, Главе № 4, а также в подразделе 5.4 Части II GMP ЕАЭС и являются гармонизированными с GMP ЕС;
— помимо указанного, на рассмотрении ЕЭК в настоящее время находится проект Руководства по обеспечению целостности данных и валидации компьютеризированных систем, в котором сформулированы детальные рекомендации относительно подходов к валидации КС и современные практические подходы к управлению целостности данных в GxP-регулируемых сферах.
Представленные в данном разделе документы относятся к так называемым «регулирующим», т. е. имеющим в той или иной степени нормативно-правовой характер. Здесь не затрагивались отдельные руководства и рекомендации, существующие на сегодняшний день в сфере GxP, в т. ч. документы международных профильных экспертных организаций, таких как, например, ISPE и PDA. О новой редакции GAMP5 – одного из ключевых руководств в данной сфере эксперты ISPE ЕАЭС уже рассказывали в одной из ранее опубликованных статей https://npjnews.com/eksperts/evrazijskoe-otdelenie-ispe-konczepcziya-pharma-4-0-ispe-novye-podhody-vo-vtoroj-redakczii-gamp-5/


