

Александр Белинский, главный консультант PQE CIS
Всепоглощающий тренд на цифровизацию «всего, что движется» часто оставляет за кадром важный аспект, касающийся того, что необходимым этапом перед началом цифровизации является предпроектное обследование и чаще всего оптимизация существующих бизнес-процессов. Ведь зачастую существующие бизнес-процессы являются противоречивыми, неоптимальными, но, так или иначе, реализуются с помощью комплекса бумажных регламентирующих и регистрирующих документов. При попытке перевода бизнес-процессов в цифровой формат серьёзно снижается эффективность их осуществления, хотя, казалось бы, цифровизация направлена на прямо противоположные цели – на повышение эффективности бизнес-процессов.
Почему так происходит? Приведу пример. В случае технологической инструкции, которая после заполнения станет протоколом производства (и элементом досье на серию), часто встречается ситуация, когда такой протокол заполняется постфактум, когда серия уже выпущена. Столкнувшись не один раз с подобными случаями, я постарался выяснить, в чём причина и обнаружил, что в форму технологической инструкции (протокола производства), состав которой предельно конкретно определен в п. 4.18 Решения № 77 ЕЭК, зачастую включается множество дополнительной информации – например, сведения о квалификационном статусе всего задействованного оборудования участка. Эта информация должна содержаться в иерархически нижестоящих по отношению к валидационному мастер-плану документах, и уж точно не рядовые операторы несут ответственность за актуальный квалификационный статус оборудования и систем. Также это может быть метрологический статус всех измерительных каналов задействованного оборудования. Хотя нет регуляторного требования именно операторам проверять в ходе рутинного производства метрологический статус (актуальность поверок или калибровок). Есть требование использовать оборудование и системы, чьи измерительные каналы имеют актуальный метрологический статус, а как именно он подтверждается – прямых ограничений нет – можно выбрать любой подходящий способ. Это вопрос процедур метрологического обеспечения. Разумеется, отвлекаясь на такие сопутствующие активности, операторы тратят массу времени, и часто оставляют заполнение протокола производства или его части «на потом» из-за занятости непосредственно производственными операциями. В то время, как есть требования по документированию выполнения шагов технологической инструкции, и оно это должно быть своевременным, иначе это, помимо всего прочего, нарушает требования в части целостности данных – пп. 4.1, 4.8.
Ниже, в Таблице 1, приведен текст оригинального GMP EU и Решения 77 ЕЭК по рассматриваемым вопросам в комплексе с требованиями по упаковке. Исходя из анализа текстов можно заключить, что формальных требований не так уже и много. Важным является уточнение, что, по сути, всю требуемую информацию можно получить, оттолкнувшись от технологической и аппаратурной схемы (хотя как раз это не требуется напрямую, но является хорошей практикой). Это позволяет детально описать все шаги в технологической инструкции (см. п. 4.18, перечисление d), чтобы иметь возможность описать подробные, постадийные инструкции, резонно иметь представление о стадиях технологического процесса; а самый удобный вариант такого представления – это как раз технологическая схема, дополненная аппаратурной схемой.
| ТЕКСТ GMP EU | ТЕКСТ РЕШЕНИЯ № 77 ЕЭК |
| Manufacturing Formula and Processing Instructions Approved, written Manufacturing Formula and Processing Instructions should exist for each product and batch size to be manufactured |
Производственная рецептура и технологические инструкции На каждый производимый лекарственный препарат и каждый размер серии необходимо иметь утвержденную письменную производственную рецептуру и технологические инструкции. |
| 4.17 The Manufacturing Formula should include: a) The name of the product, with a product reference code relating to its specification; b) A description of the pharmaceutical form, strength of the product and batch size; c) A list of all starting materials to be used, with the amount of each, described; mention should be made of any substance that may disappear in the course of processing; d) A statement of the expected final yield with the acceptable limits, and of relevant intermediate yields, where applicable |
4.17. Производственная рецептура должна включать в себя: a) наименование лекарственного препарата со ссылкой на код в соответствии со спецификацией; b) описание лекарственной формы, дозировки препарата и размера серии; c) перечень всех исходных материалов, которые будут использоваться, с указанием количества каждого. Также должны быть указаны все вещества, которые могут исчезнуть в ходе технологического процесса; d) ожидаемый выход готовой продукции с указанием допустимых пределов и выходы соответствующих промежуточных продуктов, где это возможно. |
| 4.18 The Processing Instructions should include: a) A statement of the processing location and the principal equipment to be used; b) The methods, or reference to the methods, to be used for preparing the critical equipment (e.g. cleaning, assembling, calibrating, sterilising); c) Checks that the equipment and work station are clear of previous products, documents or materials not required for the planned process, and that equipment is clean and suitable for use; d) Detailed stepwise processing instructions [e.g. checks on materials, pre-treatments, sequence for adding materials, critical process parameters (time, temp etc)]; e) The instructions for any in-process controls with their limits; f) Where necessary, the requirements for bulk storage of the products; including thecontainer, labeling and special storage conditions where applicable; g) Any special precautions to be observed. |
4.18. Технологические инструкции должны содержать: a) данные о месте осуществления процесса и об основном оборудовании, которое должно при этом использоваться; b) методы или ссылки на методы, которые должны использоваться для подготовки критического оборудования (например, очистка, монтаж, калибровка, стерилизация); c) инструкции по проверке того, что оборудование и рабочее место свободны от предыдущей продукции, ненужных для запланированного процесса документов и материалов, а также по проверке чистоты оборудования и его готовности к следующему процессу; d) подробные постадийные технологические инструкции (например, проверка материалов, предварительная обработка, порядок загрузки сырья, критические параметры процесса (время, температура и т. п.)); e) инструкции по всем видам контроля в процессе производства с указанием допустимых пределов; f) требования к хранению нерасфасованной продукции при необходимости, включая требования к таре, маркировке и специальным условиям хранения, где это требуется; g) все подлежащие соблюдению особые меры предосторожности. |
| Packaging Instructions | Инструкции по упаковке |
| 4.19 Approved Packaging Instructions for each product, pack size and type should exist. These should include, or have a reference to, the following: a) Name of the product; including the batch number of bulk and finished product; b) Description of its pharmaceutical form, and strength where applicable; c) The pack size expressed in terms of the number, weight or volume of the product in the final container; d) A complete list of all the packaging materials required, including quantities, sizes and types, with the code or reference number relating to the specifications of each packaging material; e) Where appropriate, an example or reproduction of the relevant printed packaging materials, and specimens indicating where to apply batch number references, and shelf life of the product; f) Checks that the equipment and work station are clear of previous products, documents or materials not required for the planned packaging operations (line clearance), and that equipment is clean and suitable for use. g) Special precautions to be observed, including a careful examination of the area and equipment in order to ascertain the line clearance before operations begin; h) A description of the packaging operation, including any significant subsidiary operations, and equipment to be used; i) Details of in-process controls with instructions for sampling and acceptance limits. |
4.19. Для каждого лекарственного препарата, размера и типа упаковки должны быть в наличии инструкции по упаковке. Как правило, они должны включать в себя следующие сведения (ссылки на них): a) наименование лекарственного препарата, включая номер серии нерасфасованной продукции и готового продукта; b) описание его лекарственной формы и дозировки (где применимо); c) количество лекарственного препарата в окончательной упаковке, выраженное в штуках, единицах массы или объема; d) полный перечень всех необходимых упаковочных материалов, включая их количество, размер и тип с указанием кода или номера, относящихся к спецификациям на каждый упаковочный материал; e) где применимо, образец или копия соответствующих печатных упаковочных материалов и образцы, указывающие на место нанесения номера серии и срока годности продукции; f) требования по проверке того, что оборудование и рабочее место свободны от предыдущей продукции, документов или материалов, ненужных для запланированных операций по упаковке (очистка линии), а также по проверке чистоты оборудования и его готовности к следующему процессу; g) сведения о подлежащих соблюдению специальных мерах предосторожности (включая тщательную проверку зоны упаковки и оборудования), гарантирующих очистку упаковочной линии перед началом работы; h) описание процесса упаковки со всеми основными вспомогательными операциями и используемым оборудованием; i) описание контроля в процессе производства с указаниями по отбору проб и допустимых пределов. Дополнительно могут быть разработаны иные документы, конкретизирующие положения производственной рецептуры и технологических инструкций. |
| Batch Processing Record | Записи по производству серии |
| 4.20 A Batch Processing Record should be kept for each batch processed. It should be based on the relevant parts of the currently approved Manufacturing Formula and Processing Instructions, and should contain the following information: a) The name and batch number of the product; b) Dates and times of commencement, of significant intermediate stages and of completion of production; c) Identification (initials) of the operator(s) who performed each significant step of the process and, where appropriate, the name of any person who checked these operations; d) The batch number and/or analytical control number as well as the quantities of each starting material actually weighed (including the batch number and amount of any recovered or reprocessed material added); e) Any relevant processing operation or event and major equipment used; f) A record of the in-process controls and the initials of the person(s) carrying them out, and the results obtained; g) The product yield obtained at different and pertinent stages of manufacture; h) Notes on special problems including details, with signed authorisation for any deviation from the Manufacturing Formula and Processing Instructions; i) Approval by the person responsible for the processing operations. Note: Where a validated process is continuously monitored and controlled, then automatically generated reports may be limited to compliance summaries and exception/ out-of-specification (OOS) data reports. |
4.20. На каждую произведенную серию следует сохранять записи по производству серии. Они должны основываться на соответствующих частях утвержденных документов (производственной рецептуры и технологических инструкций) и содержать следующую информацию: a) наименование и номер серии продукции; b) даты и время начала и завершения технологического процесса, а также основных промежуточных стадий; c) фамилия и инициалы оператора (операторов) каждой основной технологической операции, а также лица, проверившего каждую из этих операций, при необходимости; d) номер серии и (или) номер аналитического контроля, а также фактически отвешенное количество исходных материалов каждого вида (включая номер серии и количество любого добавленного регенерированного или переработанного материала); e) сведения о любой относящейся к делу технологической операции или любом действии, а также об основном использованном оборудовании; f) записи по контролю в процессе производства с указанием исполнителей и полученных результатов; g) выход продукции на различных стадиях производства; h) сведения об особых проблемах с подписанным разрешением на любое отклонение от технологических инструкций; i) подпись лица, ответственного за технологический процесс, с указанием даты. В случае если валидированный процесс подвергается постоянному мониторингу и контролю, автоматически создаваемые отчеты могут ограничиваться кратким резюме о соответствии отчетам об отклонениях (отступлениях) от спецификации. |
| Batch Packaging Record | Записи по упаковке серии |
| 4.21 A Batch Packaging Record should be kept for each batch or part batch processed. It should be based on the relevant parts of the Packaging Instructions. The batch packaging record should contain the following information: a) The name and batch number of the product; b) The date(s) and times of the packaging operations; c) Identification (initials) of the operator(s) who performed each significant step of the process and, where appropriate, the name of any person who checked these operations; d) Records of checks for identity and conformity with the packaging instructions, including the results of in-process controls; e) Details of the packaging operations carried out, including references to equipment and the packaging lines used; f) Whenever possible, samples of printed packaging materials used, includingspecimens of the batch coding, expiry dating and any additional overprinting; g) Notes on any special problems or unusual events including details, with signed authorisation for any deviation from the Packaging Instructions; h) The quantities and reference number or identification of all printed packaging materials and bulk product issued, used, destroyed or returned to stock and the quantities of obtained product, in order to provide for an adequate reconciliation. Where there are there are robust electronic controls in place during packaging there may be justification for not including this information; i) Approval by the person responsible for the packaging operations |
4.21. На каждую произведенную серию или часть серии следует сохранять записи по упаковке серии. Они должны основываться на соответствующих частях инструкций по упаковке. Записи по упаковке серии должны включать в себя следующие данные: a) наименование и номер серии лекарственного препарата; b) дата (даты) и время проведения операций по упаковке; c) фамилия и инициалы оператора (операторов) каждой основной технологической операции, а также лица, проверившего каждую из этих операций, при необходимости; d) записи проверок идентичности и соответствия инструкциям по упаковке, включая результаты контроля в процессе производства; e) подробные сведения об осуществленных операциях по упаковке, включая ссылки на использованное оборудование и упаковочные линии; f) образцы использованного печатного упаковочного материала, включая образцы с нанесенными номером серии, сроком годности и прочими дополнительными маркировочными данными, где применимо; g) сведения об особых проблемах или необычных происшествиях с подписанным разрешением на любое отклонение от инструкций по упаковке; h) количество и ссылка на номер или наименование всех печатных упаковочных материалов и нерасфасованной продукции, выданных, использованных, уничтоженных или возвращенных на склад, а также количество готового продукта для составления соответствующего баланса. Электронный контроль в процессе упаковки является основанием для невключения такой информации; i) подпись лица, ответственного за процесс упаковки, с указанием даты. |
Выше по тексту выделены красным цветом отсылки к тому, что объем документирования может быть заметно снижен при наличии валидированных элементов автоматизации как в части мониторинга, так и в части итогового материального баланса.
Такую же идею можно обнаружить в PDA TR № 84, который посвящён вопросам целостности данных в производственных процессах:
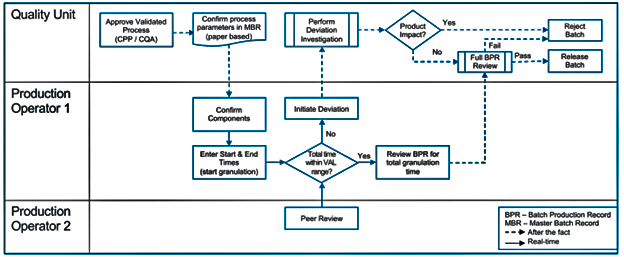
Рис. 1. Случай 1: операция гранулирования (бумажный ручной процесс без автоматизированных аварийных пределов) (PDA TR No. 84)
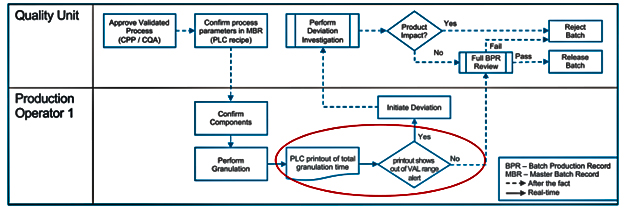
Рис. 2. Случай 2: операция гранулирования (PLC-контролируемый процесс
с автоматизированными аварийными пределами) (PDA TR No. 84)
На Рис. 1 показан случай, когда имеется бумажная технологическая рецептура и технологические инструкции (в терминологии PDA MBR – master batch record). MBR можно соотнести с технологической инструкцией, которая является по большому счету шаблоном для заполнения по мере осуществления технологического процесса. В терминологии GMP EU это регламентирующий документ, по которому должен вестись технологический процесс. А вот уже BPR – batch production record – это и есть протокол производства, регистрирующий документ. Чаще всего заполненная технологическая рецептура, содержащая конкретные данные по конкретному осуществлённому технологическому процессу с записями, согласно требованиям п. 4.20 GMP EU. Отдельно отмечу, что термины варьируются в разных документах и юрисдикциях (EMA, FDA etc.), но физический смысл прослеживается достаточно чётко. Базисный документ – регламентирующий – говорит, как нужно осуществить процесс. В итоге, по мере осуществления процесса, создаётся регистрирующий документ, содержащий записи о конкретном экземпляре процесса. Возвращаясь к Рис. 1 мы видим, что MBR бумажная и оператор 1 вручную фиксирует время начала и окончания грануляции, равно как и её параметры (на основании валидированных CQA1, CPP2). Поэтому и требуется второй оператор, подтверждающий время грануляции в рамках валидированного диапазона.
На Рис. 2 показан тот же процесс, но с элементами автоматизации, когда время (и, надо полагать, параметры) грануляции заданы в рецептуре оборудования (в PLC), то распечатка с оборудования уже будет содержать вывод о соблюдении валидированного диапазона как в части времени, так и других параметров грануляции. Тут уже второй оператор не требуется, соответственно, итоговых записей в BPR может вноситься меньше – это и есть иллюстрация примечания к п. 4.20, выделенному красным цветом.
Разумеется, если столь подробно не препарировать собственно регуляторные требования к реализации своего технологического процесса с целью последующей цифровизации регламентирующих и регистрирующих документов, сопровождающих этот процесс, то можно запрограммировать себя на неудачу. Ведь бумажные документы, даже при регламентировании процедурного контроля, можно «заполнить потом», а вот если бизнес-процесс неоптимальный жёстко прошит в цифре, то вы просто не сможете перейти на следующий этап, пока не заполните всю информацию, в т. ч. и потенциально избыточную – как выше в отношении квалификационного или метрологического статуса, а это просто может блокировать производство и приводить вместо ожидаемой выгоды к затратам на его цифровую реализацию.

Владимир Орлов Директор Евразийского отделения ISPE
Оксана Пряничникова Заместитель директора Евразийского отделения ISPE
Сайт: www.ispe.ru
E-mail: ispe@ispe.ru


