Дисклеймер: по некоторым вопросам мнение экспертов экспертного учреждения может отличаться от предлагаемой автором трактовки.
Для того, чтобы правильно «поставить запятую», на мой взгляд, целесообразно использовать риск-ориентированный подход к оценке изменений, в частности, и обеспечению соответствия нормативным требованиям при регистрации и производстве лекарственных средств, в целом.
Перед любой производственной площадкой как минимум однажды стояла задача замены поставщиков фармацевтических субстанций, вспомогательных веществ, упаковочных материалов, реактивов и стандартных образцов, расходных материалов, технологического и аналитического оборудования.
В данной статье будут рассмотрены наиболее распространенные изменения различных аспектов производства и качества лекарственного препарата (ЛП), необходимость оценки и имплементации которых возникает неоднократно в процессе коммерческой фазы его жизненного цикла.
Все изменения будут рассматриваться со следующих позиций:
1. Повлечет ли изменение, которое планируется к имплементации, необходимость актуализации регистрационного досье;
2. На каком этапе управления изменением необходимо информировать регулятора посредством внесения изменения (в случае, если было принято решение об актуализации регистрационного досье).
Классификация изменений, сроки, в которые необходимо информировать регулятора, а также длительность и характер экспертизы будут представлены на основе нормативной парадигмы ЕАЭС, в частности Приложения 19 к Правил регистрации и экспертизы лекарственных средств для медицинского применения, утв. решением Совета ЕЭК от 03.11.2016 N78 (ред. от 23.09.2022)1.
ЗАДАЧА 1. ЗАМЕНА ПРОИЗВОДИТЕЛЯ ВСПОМОГАТЕЛЬНОГО ВЕЩЕСТВА (ВВ), ПРИ КОТОРОМ:
1.1. Новый производитель ВВ (производитель 2) осуществляет контроль качества по спецификации, которая полностью соответствует спецификации предыдущего производителя ВВ (производитель 1).
1.2. Новый производитель ВВ (производитель 2) осуществляет контроль качества по спецификации, которая не соответствует спецификации предыдущего производителя ВВ (производитель 1) по 1 или нескольким показателям/нормам/методам.
1.3. Новый производитель ВВ (производитель 2) осуществляет контроль качества по спецификации, которая полностью отличается от спецификации предыдущего производителя ВВ (производитель 1) (например, производитель 2 проводит контроль по ГОСТ, производитель 1 по ЕР).
Необходимо оценить требуется ли внесение изменений в регистрационное досье ЛП.
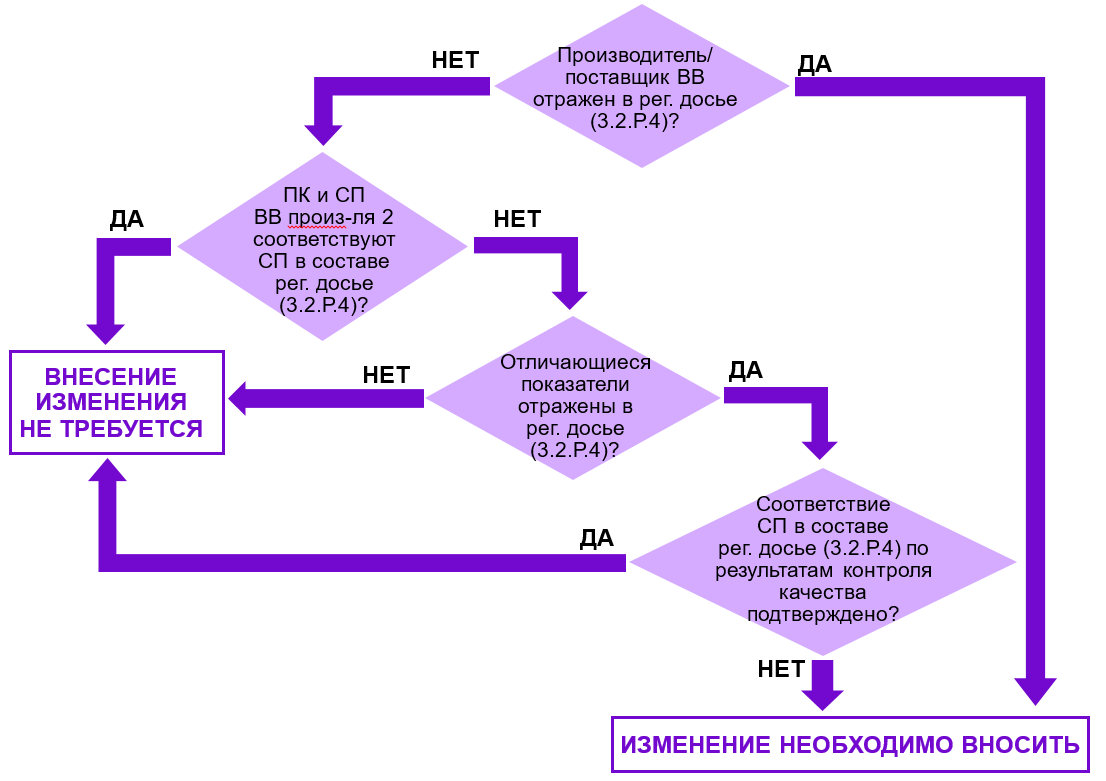
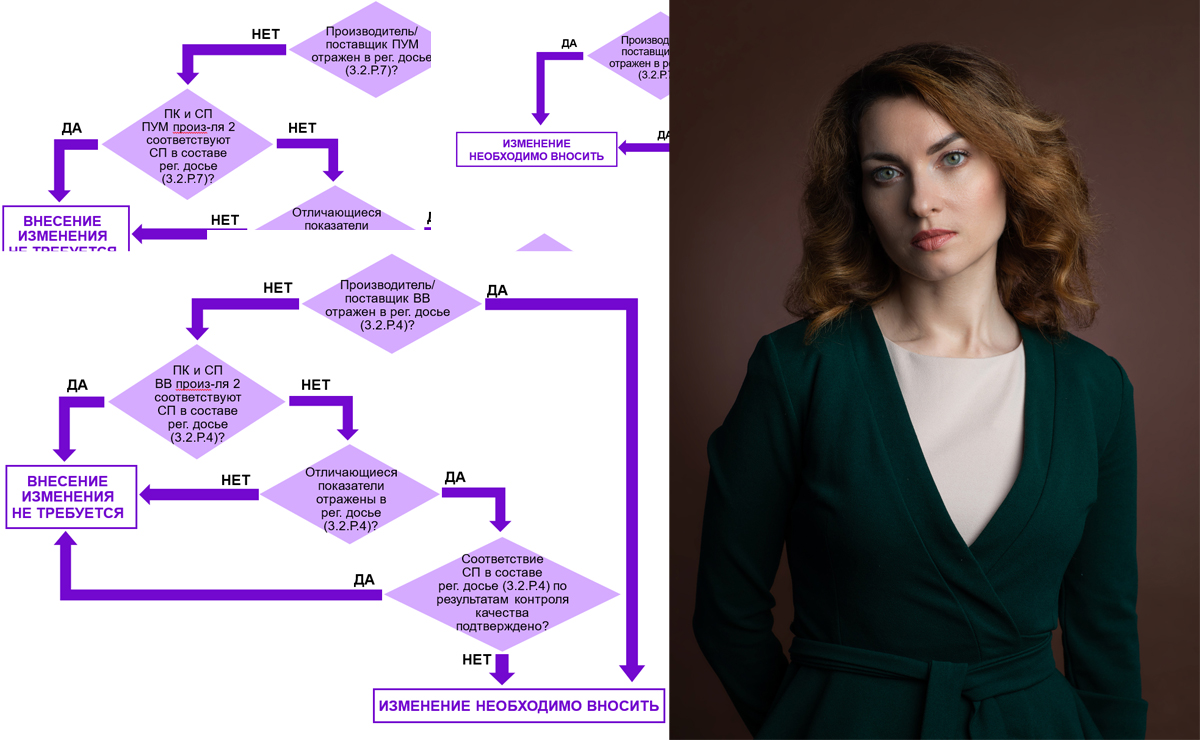
Рис. 1. Алгоритм принятия решения о необходимости внесения изменений в регистрационное досье при замене производителя вспомогательного вещества.
РЕШЕНИЕ.
Для принятия решения предлагаю воспользоваться алгоритмом, представленным на рис. 1.
Если производитель ВВ указан в регистрационном досье, что возможно только в случае, если в составе досье подавалась заводская спецификация (!), и он меняется, использование ВВ от нового производителя будет нарушением. В таком случае обязательно нужно вносить изменение.
Если производитель не заявлен в регистрационном досье, а дана ссылка на Перечень утвержденных поставщиков, то при оценке необходимости отражения данного изменения в досье нужно исходить из сравнения спецификации на ВВ в составе рег. досье (3.2.Р.4.1) и заводской спецификации.
Если документы входного контроля и/или документы по выпускающему контролю производителя соответствуют спецификации в составе регистрационного досье (3.2.Р.4.1), то основания для внесения изменений отсутствуют. В рамках плановых регистрационных мероприятий предоставляются актуальные документы по качеству ВВ, в том числе от новых производителей, что не считается изменением и не требует заявления. В свою очередь, изменения в Перечень утвержденных поставщиков вносятся в рамках контроля изменений.
Если результаты входного контроля (в том числе с привлечением контрактных лабораторий) и/или документы по выпускающему контролю производителя не позволяют подтвердить соответствие спецификации в составе регистрационного досье (3.2.Р4.1), необходимо актуализировать спецификацию путем заявления соответствующих изменений.
Согласно Правилам, если изменение предполагает ужесточение критериев приемлемости спецификации, добавление (или исключение) в спецификацию нового параметра спецификации и соответствующего ему метода испытаний, оно может быть классифицировано как незначимое изменение (тип IА). Для данных изменений предусмотрен уведомительный характер, т. е. допускается уведомить регулятора в течение 365 дней с момента его имплементации. Для данного типа изменений не предусмотрены запросы и фармацевтическая экспертиза. Экспертиза данного типа может занять от 20 до 40 р. д.2
Изменение, выходящее за одобренные критерии приемлемости спецификаций или предполагающее исключение параметра спецификации, который может существенно повлиять на совокупное качество ЛП, классифицируется как значимое изменение (тип II). Значимые изменения не могут быть имплементированы до утверждения их регулятором. Для данного типа изменений предусмотрены запрос и фармацевтическая экспертиза. Экспертиза данного типа изменений длится от 40 до 60 р. д.
Добавление или замена параметра спецификации3 и соответствующего ему метода испытаний из соображений безопасности или качества, а также добавление (или изменение) ссылки на любую фармакопею кроме Фармакопеи Союза, расценивается как изменение (тип IB). Для данных изменений не предусмотрен уведомительный характер, т. е. информация об изменении должна быть предоставлена на экспертизу до их имплементации. Но, если в течение 30 дней не было получено отрицательное решение, изменение считается одобренным. Для данного типа изменений не предусмотрены запросы, но может быть потребоваться фармацевтическая экспертиза. Экспертиза данного типа может занять от 20 до 40 р. д.
ЗАДАЧА 2. ЗАМЕНА ПРОИЗВОДИТЕЛЯ ПЕРВИЧНОГО УПАКОВОЧНОГО МАТЕРИАЛА (ПУМ), ПРИ КОТОРОМ:
2.1. Новый производитель ПУМ (производитель 2) осуществляет контроль качества по спецификации, которая полностью соответствует спецификации предыдущего производителя ПУМ (производитель 1).
2.2. Новый производитель ПУМ (производитель 2) осуществляет контроль качества по спецификации, которая не соответствует спецификации предыдущего производителя ПУМ (производитель 1) по 1 или нескольким показателям/нормам/методам.
2.3. Новый производитель ПУМ (производитель 2) осуществляет контроль качества по спецификации, которая полностью отличается от спецификации предыдущего производителя ПУМ (производитель 1) (например, производитель 2 проводит контроль по ГОСТ, производитель 1 по ЕР).
Необходимо оценить требуется ли внесение изменений в регистрационное досье ЛП.
РЕШЕНИЕ.
Для принятия решения предлагаю воспользоваться алгоритмом, представленным на рис. 2.
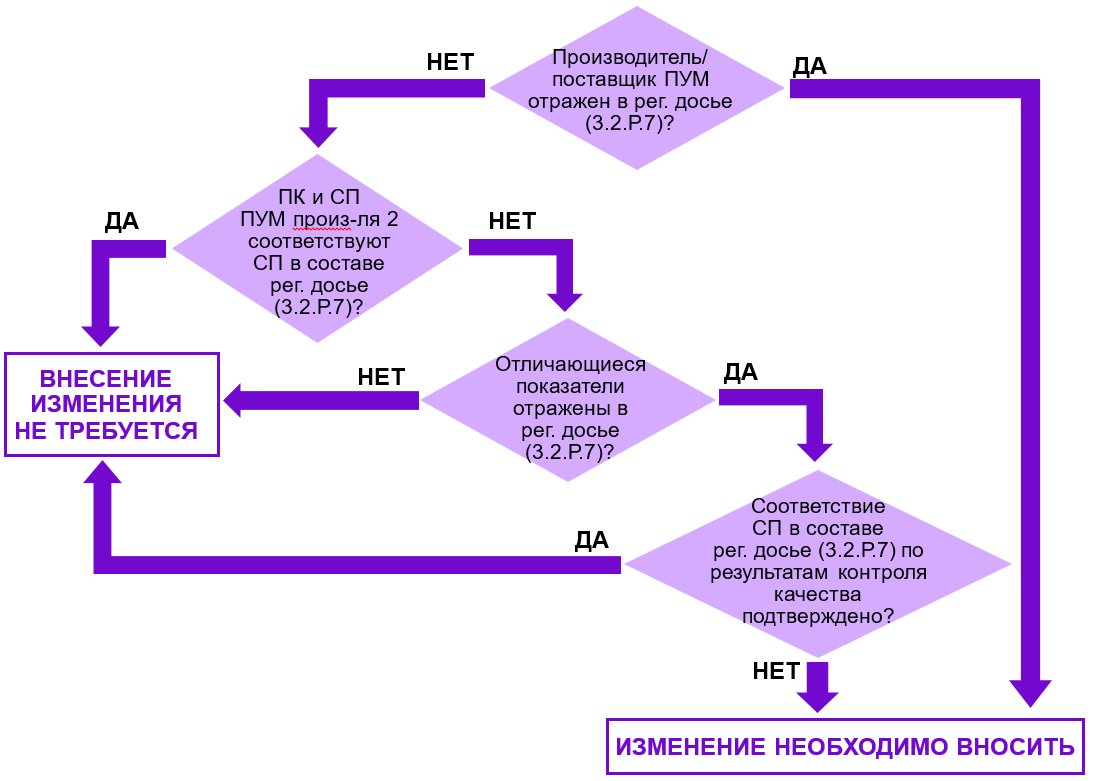
Рис. 2. Алгоритм принятия решения о необходимости внесения изменений в регистрационное досье при замене производителя первичного упаковочного материала.
Если производитель ПУМ указан в регистрационном досье, что возможно только в случае, если в составе рег. досье подавалась заводская спецификация (!), и он меняется, использование ПУМ от нового производителя будет нарушением. В таком случае нужно обязательно вносить изменение.
Если производитель не заявлен в регистрационном досье, а дана ссылка на Перечень утвержденных поставщиков, то при оценке необходимости отражения данного изменения в досье нужно исходить из сравнения спецификации на ПУМ в составе рег. досье (3.2.Р.7.1) и заводской спецификации.
Если документы входного контроля и/или документы по выпускающему контролю производителя соответствуют спецификации в составе рег. досье (3.2.Р.7.1), то основания для внесения изменений отсутствуют. В рамках плановых регистрационных мероприятий предоставляются актуальные документы по качеству ПУМ, в том числе от новых производителей, что не считается изменением и не требует заявления. В свою очередь, изменения в Перечень утвержденных поставщиков вносятся в рамках контроля изменений.
Если результаты входного контроля (в том числе с привлечением контрактных лабораторий) и/или документы по выпускающему контролю производителя не позволяют подтвердить соответствие спецификации в составе рег. досье (3.2.Р.7.1), необходимо актуализировать спецификацию путем заявления соответствующих изменений.
Согласно Правилам, любое изменение, связанное с материалом первичной упаковки ЛП, расценивается как незначимое изменение (тип IA или IВ). Экспертиза данных типов изменений занимает от 20 до 40 р. д.
Причем изменения, обусловленные добавлением или заменой параметра спецификации из соображений безопасности или качества, расцениваются как изменения типа IВ. Для данных изменений не предусмотрен уведомительный характер, т. е. информация об изменениях должна быть предоставлена на экспертизу до их имплементации. Но, если в течение 30 дней не было получено отрицательное решение, изменение считается одобренным. Для данного типа изменений не предусмотрены запросы, но может потребоваться фармацевтическая экспертиза.
Все остальные изменения, связанные с заменой производителя первичного упаковочного материала (изменение поставщика компонентов упаковки или устройства (добавление/исключение/замена); изменение параметров спецификации и/или критериев приемлемости, и/или изменение аналитической методики первичной упаковки ЛП) классифицируются как изменения типа IА. Для данных изменений предусмотрен уведомительный характер, т. е. допускается уведомить регулятора в течение 365 дней с момента его имплементации. Для данного типа изменений не предусмотрены запросы и фармацевтическая экспертиза.
ЗАДАЧА 3. ЗАМЕНА ПРОИЗВОДИТЕЛЯ ВТОРИЧНОГО УПАКОВОЧНОГО МАТЕРИАЛА (ВУМ), ПРИ КОТОРОЙ:
3.1. Новый производитель ВУМ (производитель 2) осуществляет контроль качества по спецификации, которая полностью соответствует спецификации предыдущего производителя ВУМ (производитель 1).
3.2. Новый производитель ВУМ (производитель 2) осуществляет контроль качества по спецификации, которая не соответствует спецификации предыдущего производителя ВУМ (производитель 1) по 1 или нескольким показателям/нормам/методам.
3.3. Новый производитель ВУМ (производитель 2) осуществляет контроль качества по спецификации, которая полностью отличается от спецификации предыдущего производителя ВУМ (производитель 1) (например, производитель 2 проводит контроль по ГОСТ, производитель 1 по ЕР).
Необходимо оценить требуется ли внесение изменений в регистрационное досье ЛП.
РЕШЕНИЕ.
Для принятия решения предлагаю воспользоваться алгоритмом, представленным на рис. 3.
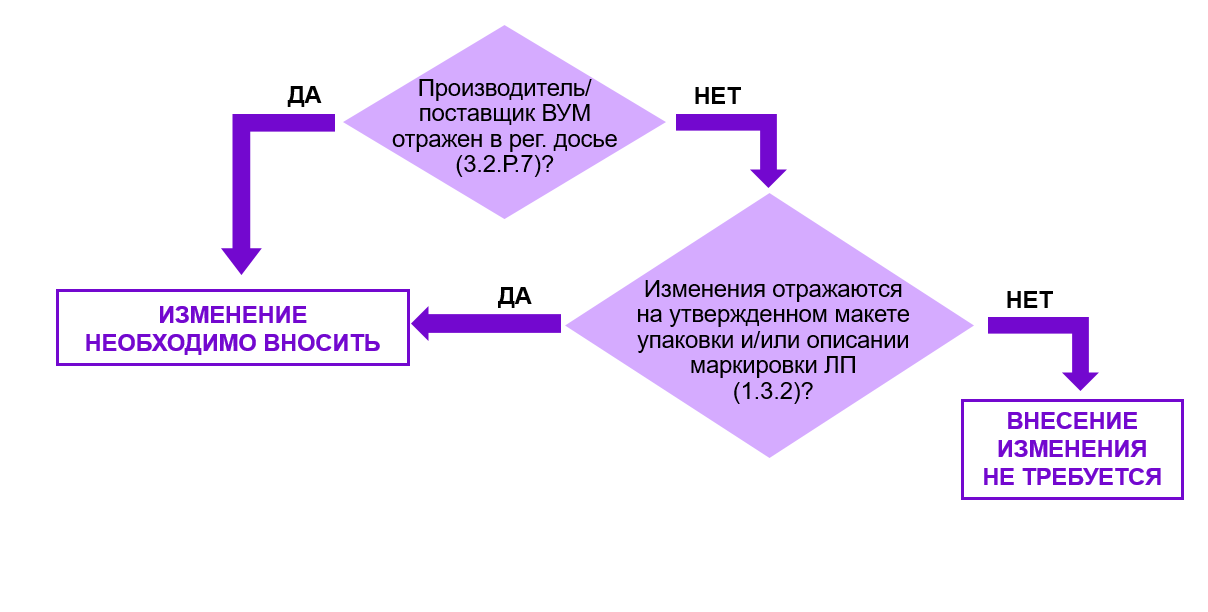
Рис. 3. Алгоритм принятия решения о необходимости внесения изменений в регистрационное досье при замене производителя вторичного упаковочного материала.
Поскольку вторичный упаковочный материал не оказывает влияния на качество ЛП, регулятора он интересует только с точки зрения отражения на нем информации, необходимой для идентификации ЛП и его безопасного применения.
В связи с чем, для принятия решения о необходимости внесения изменений в регистрационное досье в случае замены производителя вторичного упаковочного материала необходимо принять во внимание 2 фактора.
Во-первых, оценить степень соответствия утвержденного макета упаковки и макета упаковки от нового производителя. Степень соответствия во многом зависит от степени детализации и формата информации о макете упаковки. Согласно требованиям к маркировке4 отсутствует необходимость отражения размеров первичной/вторичной упаковки (или самоклеящейся этикетки/ стикера и т. д.) и формулы цветового решения на макетах в составе регистрационного досье. Но в большинстве случаев, одна, как правило формула цветового решения, а иногда и обе эти характеристики отражаются на макете, подаваемом в рамках регистрационных процедур. Очевидно, что чем выше степень детализации информации о макете, тем выше вероятность возникновения необходимости его актуализации при изменениях, сопряженных с заменой производителя материала вторичной упаковки.
Во-вторых, необходимо оценить, отражен ли в регистрационном досье производитель вторичного упаковочного материала. Если производитель ВУМ указан в рег. досье, что возможно только в случае, если в рег. досье подавалась заводская спецификация (!), и он меняется, использование ВУМ от нового производителя будет нарушением. В таком случае необходимо вносить изменение.
Если производитель не заявлен в рег. досье, а дана ссылка на Перечень утвержденных поставщиков, то при оценке необходимости отражения данного изменения в рег. досье нужно исходить из сравнения спецификации на ВУМ в составе рег. досье (3.2.Р.7.1) и заводской спецификации.
Если документы входного контроля и/или документы по выпускающему контролю производителя соответствуют спецификации в составе рег. досье (3.2.Р.7.1), то основания для внесения изменений отсутствуют. В рамках плановых регистрационных мероприятий предоставляются актуальные документы по качеству ВУМ, в том числе от новых производителей, что не считается изменением и не требует заявления. В свою очередь, изменения в Перечень утвержденных поставщиков вносятся в рамках контроля изменений.
Если результаты входного контроля (в том числе с привлечением контрактных лабораторий) и/или документы по выпускающему контролю производителя не позволяют подтвердить соответствие спецификации в составе регистрационного досье (3.2.Р7.1), необходимо актуализировать спецификацию путем заявления соответствующих изменений.
Однако, этот вариант развития событий можно практически исключить, если руководствоваться нормативными требованиями. Согласно Правилам5, для нефункциональных материалов вторичной (потребительской) и промежуточной упаковки (например, не обеспечивающих дополнительную защиту и не доставляющих ЛП) достаточно представить только краткое описание. Для функциональных компонентов вторичной (потребительской) и промежуточной упаковки представляется дополнительная информация. Таким образом, рационально стремиться к оптимально-достаточному представлению информации в зависимости от ее прямых целей и задач и не добиваться эквивалентности содержания спецификации, отраженной в разделе 3.2.Р.7.1 рег. досье, и заводской спецификации. Поскольку для первой достаточно меньшее количество показателей. Поэтому совершенно законно и целесообразно представлять информацию о материалах вторичной упаковки в составе рег. досье без технически обусловленных показателей (а, соответственно, без норм и методов контроля этих показателей), которые отражаются в заводской спецификации. В противном случае ДРУ обречен на бесконечный процесс внесения изменений в регистрационное досье или выпуск ЛП не в соответствии с утвержденным рег. досье, т. е. с нарушением требований GMP (допускается выбрать приемлемый для себя вариант :)).
Вместе с тем, согласно Правилам, все изменения, связанные с материалом вторичной упаковки ЛП, расцениваются как незначимые изменения (тип IA). Экспертиза данного типа изменений занимает от 20 до 40 р. д.
Причем, если изменение, связанное с заменой производителя вторичного упаковочного материала, затрагивает информацию о ЛП (т. е. отражается на макете вторичной упаковки (например, изменение дизайна, цвета маркировки, нанесение штрих кода (2D, 3D), нанесение шрифта Брайля. Данные изменения не могут быть имплементированы до их утверждения регулятором. Для данного типа изменений не предусмотрены запросы и фармацевтическая экспертиза.
Все остальные изменения, связанные с заменой производителя вторичного упаковочного материала (изменение поставщика компонентов упаковки или устройства (добавление/исключение/замена) (при условии его отражения в материалах регистрационного досье) и другие изменения какой-либо составляющей упаковочного материала, непосредственно не соприкасающейся с ЛП (например, цвет съемных колпачков, цветные кодовые кольца на ампулах, изменение колпачка, защищающего иглу (использование другого пластика) и т. д.) классифицируются как изменения типа IА. Для данных изменений предусмотрен уведомительный характер, т. е. допускается уведомить регулятора в течение 365 дней с момента их имплементации. Для данного типа изменений не предусмотрены запросы и фармацевтическая экспертиза.
ЗАДАЧА 4. ЗАМЕНА РЕАКТИВА, ИСПОЛЬЗУЕМОГО В КОНТРОЛЕ ЛП
4.1. Измененный реактив фармакопейный.
4.2. Измененный реактив нефармакопейный.
Необходимо оценить, требуется ли внесение изменений в регистрационное досье ЛП.
РЕШЕНИЕ.
Для принятия решения предлагаю воспользоваться алгоритмом, представленным на рис. 4.
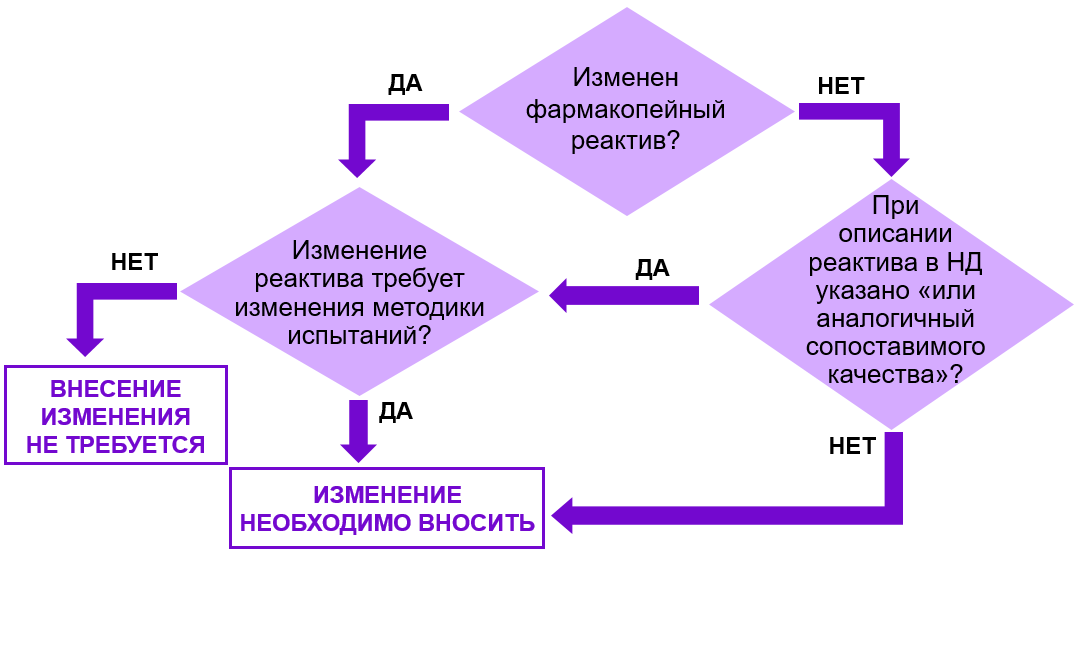
Рис. 4. Алгоритм принятия решения о необходимости внесения изменений в регистрационное досье при замене реактива, используемого для контроля качества лекарственного препарата.
Для принятия решения о необходимости внесения изменений в регистрационное досье в случае изменения реактива необходимо принять во внимание каким образом информация об этом реактиве отражена в НД и требует ли замена реактива изменения методики испытаний. Причем неизменность методики и возможность использования нового реактива должна быть подтверждена результатами ее валидации6. Равно как и степень соответствия замененного реактива ранее заявленному должна быть оценена на основании соответствующих данных (сертификатов качества), подтверждающих его пригодность заявленным целям.
В противном случае требуется вносить изменения в регистрационное досье.
В зависимости от типа методики испытаний и ее значимости при оценке качества препарата данное изменение, согласно Правилам, может быть классифицировано как незначимое изменение типа IA или IB. Экспертиза данного типа изменений занимает от 20 до 40 р. д., но в случае с изменением типа IB может быть назначена фармэкспертиза.
ЗАДАЧА 5. ЗАМЕНА ТЕХНОЛОГИЧЕСКОГО ОБОРУДОВАНИЯ, ЕСЛИ
5.1 коутер/смеситель аналогичный по характеристикам ранее использованному;
5.2. коутер/смеситель иного объема в отличие от ранее использованного.
Необходимо оценить требуется ли внесение изменений в регистрационное досье ЛП.
РЕШЕНИЕ.
Для принятия решения предлагаю воспользоваться алгоритмом, представленным на рис. 5.
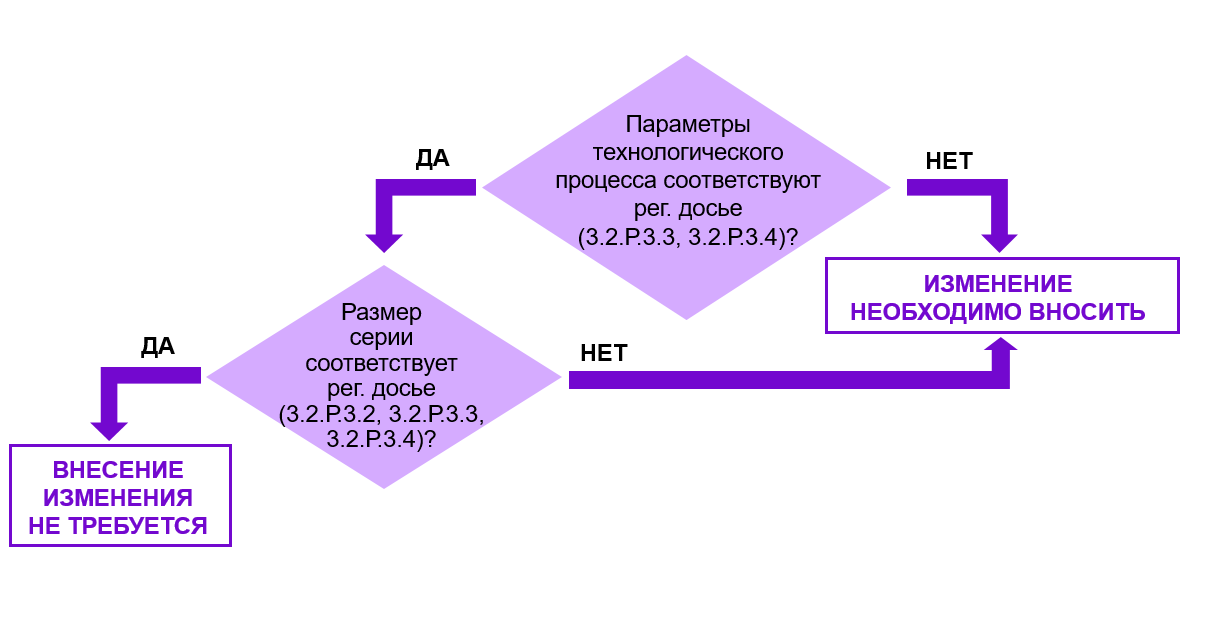
Рис. 5. Алгоритм принятия решения о необходимости внесения изменений в регистрационное досье при замене технологического оборудования.
Для принятия решения о необходимости внесения изменений в регистрационное досье в случае замены технологического оборудования необходимо принять во внимание 2 фактора.
Во-первых, необходимо оценить влияет ли замена технологического оборудования на параметры технологического процесса, отраженные в разделах 3.2.Р.3.3 и 3.2.Р.3.4.
Во-вторых, необходимо оценить, приводит ли замена технологического оборудования к изменению объёма(ов) серии(й).
Если параметры процесса и объем серии остаются неизменными, то основания для внесения изменений отсутствуют.
При изменении заявленных параметров технологического процесса и/или объемов серий, требуется актуализация регистрационного досье.
Согласно Правилам, все изменения, связанные с заменой технологического оборудования, используемого при производстве ЛП в зависимости от их характера и степени влияния на качество, безопасность и эффективность ЛП, могут быть классифицированы как незначительные изменения типа (IА или IB) или значимые изменения (типа II). Более детально информация по возможным кодам и типам изменений представлена в таблице 1.
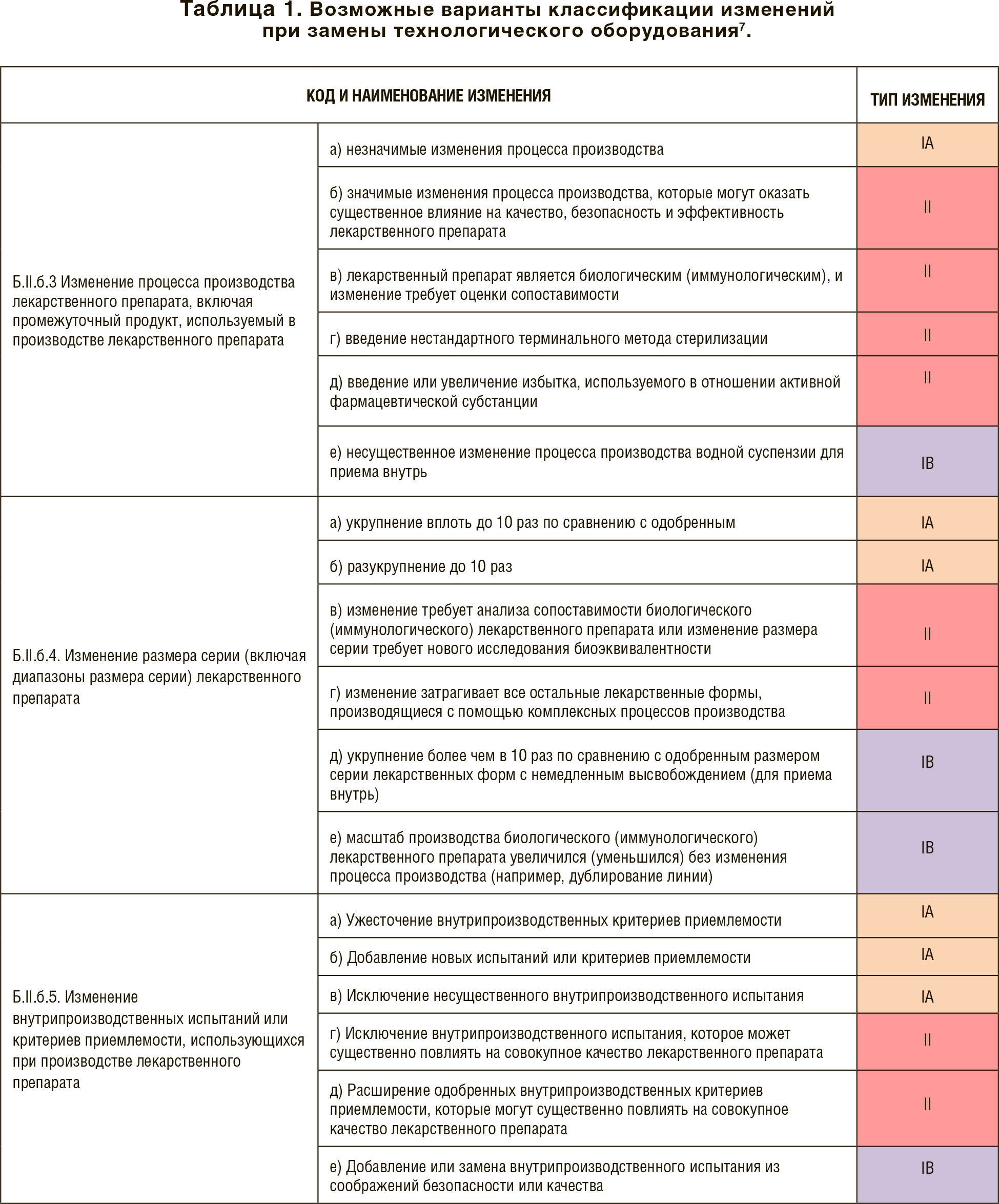
В зависимости от типа изменений варьирует характер информирования регуляторного органа, продолжительность экспертизы (от 20 до 60 р. д.), возможность получения запросов и необходимость проведения фармэкспертизы.
ЗАДАЧА 6. ЗАМЕНА ФАСОВОЧНО-УПАКОВОЧНОЙ ЛИНИИ, ЕСЛИ
6.1. фасовочно-упаковочная линия аналогична по характеристикам ранее использованной;
6.2. фасовочно-упаковочной линия отлична по характеристикам от ранее использованной.
Необходимо оценить требуется ли внесение изменений в регистрационное досье ЛП.
РЕШЕНИЕ.
Для принятия решения предлагаю воспользоваться алгоритмом, представленным на рис. 6.
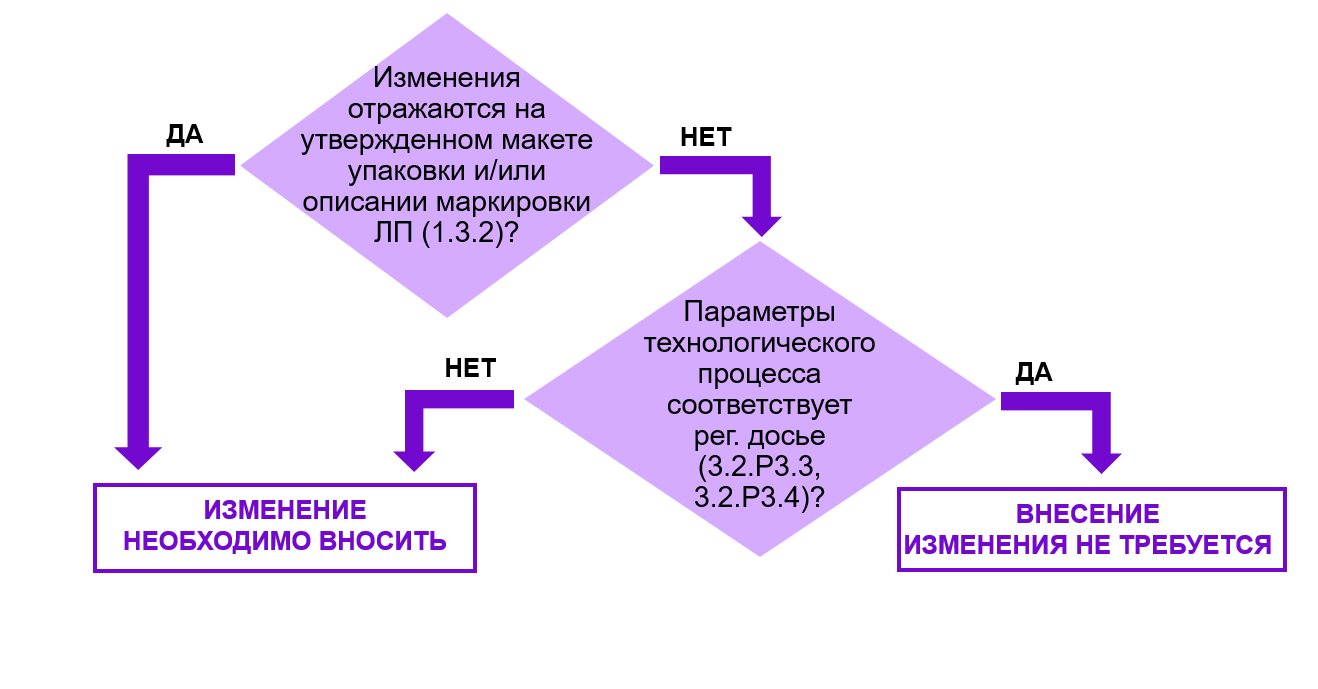
Рис. 6. Алгоритм принятия решения о необходимости внесения изменений в регистрационное досье при замене фасовочно-упаковочного оборудования.
В случае замены фасовочно-упаковочного оборудования рассуждения в целом будут аналогичны тем, что были при замене технологического оборудования.
Для принятия решения о необходимости внесения изменений в регистрационное досье необходимо принять во внимание 2 фактора.
Во-первых, необходимо оценить отражается ли замена фасовочно-упаковочного оборудования на макете упаковки и/или описании маркировки (1.3.2).
Во-вторых, необходимо оценить сопровождается ли замена фасовочно-упаковочного оборудования изменением параметров технологического процесса, которые отражены в разделах 3.2.Р.3.3 и 3.2.Р.3.4.
Если макет упаковки, описание маркировки (1.3.2) и/или параметры процесса остаются неизменными, то основания для внесения изменений отсутствуют.
При изменении макета упаковки, описания маркировки (1.3.2) и/или параметров технологического процесса, отраженных в регистрационном досье, требуется актуализация последнего.
При необходимости внесения изменений, обусловленных заменой фасовочно-упаковочной линии, возможны следующие варианты.
Если замена фасовочно-упаковочной линии отразилась на макете упаковки, такое изменение, согласно Правилам, классифицируется как незначимое изменение типа IАну. Характеристика данного типа изменения дана мною выше в решении задачи 3 «Замена производителя вторичного упаковочного материала».
Если замена фасовочно-упаковочной линии не отразилась на макете упаковки, но сопровождается изменением параметров процесса фасовки/ упаковки, такое изменение можно классифицировать как незначимое изменение типа IА. Характеристика данного типа изменений уже неоднократно давалась мною выше.
При классификации изменений необходимо принимать во внимание, что любое изменение может иметь несколько следствий, соответственно, необходимо будет присвоить код каждому из них. В этом случае, данные изменения (по сути следствия) однозначно могут быть сгруппированы в одном заявлении, поскольку являются связанными, при этом возможные сроки имплементации определяются по так называемому «наихудшему случаю».
ЗАДАЧА 7. ЗАМЕНА ПРОИЗВОДИТЕЛЯ АФС, ПРИ КОТОРОЙ:
7.1. Новый производитель АФС (производитель 2) осуществляет контроль качества по спецификации, которая полностью соответствует спецификации предыдущего производителя АФС (производитель 1).
7.2. Новый производитель АФС (производитель 2) осуществляет контроль качества по спецификации, которая не соответствует спецификации предыдущего производителя АФС (производитель 1) по 1 или нескольким показателям/нормам/методам и/или упаковке или условиям хранения.
Необходимо оценить требуется ли внесение изменений в регистрационное досье ЛП.
РЕШЕНИЕ.
Замена производителя АФС в любой ситуации не может остаться «незамеченной» и требует одобрения регулятора до его внедрения. При всех указанных условиях данное изменение классифицируется как значимое изменение (тип II), которое требует проведения фармацевтической экспертизы. При этом, если новый производитель имеет сертификат соответствия Европейской Фармакопее, данное изменение классифицируется как незначительное изменение типа IАну (код Б.III.1а), которое может быть подано одновременно с досье на приведение в соответствие. В таком случае, данное изменение может быть утверждено без проведения фамэкспертизы.
ЗАКЛЮЧЕНИЕ
В заключение еще раз подчеркну, что решение о необходимости внесения изменений в регистрационное досье зависит от той информации, которая в нем отражена, и целесообразно не слепо следовать выполнению нормативных требований, а принимать его в альянсе между подразделениями качества и регистрации на основе оценки рисков для обеспечения соответствия нормативным требованиям при регистрации и производстве ЛП. Применение риск-ориентированных подходов к оценке пострегистрационных изменений аспектов производства и качества свидетельствует об осознанности и высоком уровне развития системы качества ДРУ и производителя, что, в свою очередь, позволяет предоставить неформальные гарантии обеспечения потребителей качественными ЛП с максимально возможным уровнем эффективности и безопасности и, как следствие, благоприятным соотношением «польза-риск».
Надеюсь, что представленные в статье алгоритмы помогут в принятии решений о необходимости отражения изменений по производству и качеству в регистрационном досье.
Данная статья является частью проекта «Регуляторная осознанность». Другие материалы проекта доступны на сайте: https://irina-krasnova.ru/


