
Юрий Сандлер, Генеральный директор PQE CIS

Александр Белинский, Главный консультант PQE CIS
Данная статья подготовлена при участии Евразийского отделения ISPE
В рамках развития образовательного направления ISPE ЕАЭС и углубления сотрудничества с «Евразийской Академией надлежащих практик» при участии экспертов PQE Group в феврале 2022 года состоялся вебинар для фармпроизводителей ЕАЭС «Валидация технологического процесса – подход на основании жизненного цикла продукта. Концепция Quality by Design – качество через дизайн». В вебинаре приняли участие: Владимир Орлов, директор Евразийского отделения ISPE, Юрий Сандлер, генеральный директор PQE CIS, Александр Белинский, Главный консультант PQE CIS. В программе были освещены такие вопросы, как:
- Концепция Quality by Design – качество посредством проектирования. Обзор руководств ISPE и практические аспекты их применения, включая руководство о жизненном цикле валидации процесса (ISPE Good Practice Guide: Practical Implementation of the Lifecycle Approach to Process Validation), которое было представлено на Ежегодной конференции ISPE ЕАЭС в ноябре 2021 г.
- Что необходимо выполнить в ходе фармацевтической разработки. Необходимо установить критические показатели качества (CQA) и определяемые ими критические параметры процесса (СPP), критические характеристики материалов (CMA). Рекомендуется (как удобная опция) определять пространство проектных параметров (Design Space).
- Что делать производителю с уже производимой лекарственной продукцией для рынка ЕАЭС? Ведь даже при наличии валидации процесса по традиционному подходу для них обязательной становится продолжающаяся верификация процесса в ходе жизненного цикла.
- Современные подходы к валидации технологических процессов. В данной статье приводится краткое содержание вебинара, которое раскрывает читателям подход и мнение экспертов ISPE и PQE по данной тематике.
Quality by Design – есть у валидации начало, нет у валидации конца.
Слоган выше, характеризующий валидацию, зачастую на различных конференциях произносят по-философски с некоторым налётом грусти. До недавнего времени – без «диковинного» словосочетания «Quality by Design» – «качество через дизайн». Но в связи с вступлением силу обновленного Приложения 15 GMP ЕАЭС (в форме Решения Совета Евразийской экономической комиссии от 14 июля 2021 г. № 65 «О внесении изменений в Правила надлежащей производственной практики Евразийского экономического союза») к этим тяжким раздумьям добавилось и новое понятие, хотя новизной является только его официально закрепленная возможность применения. Вместе с тем, если пойти немного вглубь, то в рамках ЕАЭС концепция «качество через дизайн» впервые прозвучала в тексте Рекомендации Коллегии Евразийской экономической комиссии от 26 сентября 2017 г. № 19 «О Руководстве по валидации процесса производства лекарственных препаратов для медицинского применения». Не говоря уже о том, что и Решение № 65 (соответствующее в целом приложению 1 5 GMP EU) и Рекомендация № 19 (соответствующее в целом Руководству ЕМА по валидации процесса) всего лишь актуализируют достаточно давно прорабатываемый концепт, сменяющий подход «Quality by Testing», как неприемлемый в виду запредельной контрпродуктивности. Делая акцент на тестирование готовой продукции, мы пытались компенсировать отсутствие знаний о продукте и процессе, что приводило к значительному риску производства некачественного продукта и убыткам для компании.
Что же скрывается за словосочетанием «качество через дизайн» (Quality by Design), которое теперь предложено к рассмотрению? Оттолкнемся от определения, данного по текстам Решения 65 и Рекомендации № 19 – это системный подход, предусматривающий определение целей перед началом разработки продукции, точную оценку продукции и процесса ее производства, а также контроль процесса производства на основе научных данных и принципов управления рисками для качества.
Звучит это всё как «пугающее наукообразие». Мало того, что валидация процесса на современном этапе перестает быть разовым событием – уже исключен вариант произвести «коекак» три валидационные серии и «забыть» о валидации на какое-то время – так вдобавок ещё и предлагается осуществлять контроль процесса производства на основе научных подходов. Таким образом, наукоемкость и ресурсоемкость процесса очевидно возрастают. При этом не следует забывать, что если подход «качество через дизайн» является в настоящее время допустимой альтернативой, то продолжающаяся верификация процесса в ходе жизненного цикла становится безальтернативным обязательством.
Казалось бы, эти нововведения могут восприниматься как очередной «регуляторный прессинг», «гиря на ногах бизнеса». Однако при более детальном рассмотрении выясняется, что как раз реализация концепта «качество через дизайн» способна сократить затраты по выведению на рынок новых лекарственных препаратов вплоть до 25%. За счет чего может быть достигнут такой эффект? По сравнению с традиционным подходом (когда этап фарм– разработки выполнялся без должного статистического обоснования, во многом эмпирическим путем, «по наитию»), больший фокус внимания уделяется именно этапу фармацевтической разработки – именно там формируется целевой профиль продукта, а также связанные с ним CQA (критические показатели качества), CPP (критические параметры процесса), как возможная опция – пространство проектных параметров (Design Space) и стратегия контроля. Считается, что реализация такого концепта не принесёт какой-то значимой экономии на этапе фармразработки, однако обеспечит статистически обоснованные знания о продукте и процессе, что заметно упростит масштабирование, трансфер и производство последующих коммерческих серий, поскольку процесс будет заведомо в состоянии статистического контроля (см. рис. 1).
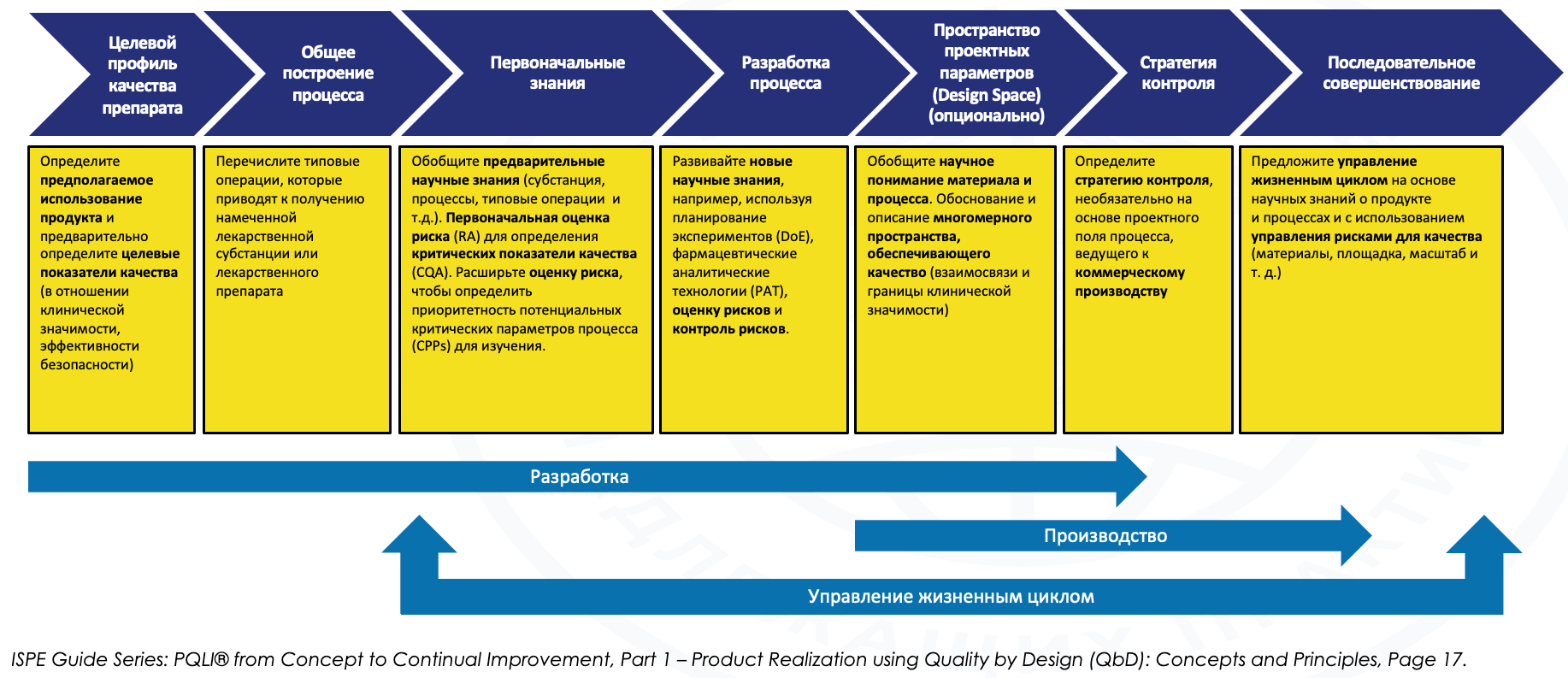
Рис. 1. Обзор концепта «качество через дизайн»
Далее следует краткое описание главных этапов процесса при разработке лекарственных средств согласно ICH Q8 и руководствам PQLI от ISPE от ранней разработки до производства коммерческих серий.
Этапы расположены в логическом и хронологическом порядке:
- Определение целевого профиля качества препарата (Quality Target Product Profile, QTPP) – Задача данного этапа обеспечить понимание того, что будет гарантировать качество, безопасность и эффективность конкретного продукта для пациента.
QTPP описывает критерии проекта для продукта, формирует основу для разработки CQA, CPP и стратегии контроля.
Аспекты целевого профиля качества продукта могут включать, но не ограничиваются следующим:
Предполагаемое применение в клинической практике, способ введения, лекарственная форма, системы доставки;
- Дозировка (–и);
- Система упаковки/укупорки;
- Высвобождение или доставка лекарственного вещества и свойства, влияющие на фармакокинетические характеристики (например, растворение, всасывание и т. д.), которые соответствуют разрабатываемой лекарственной форме препарата;
- Критерии качества лекарственного препарата (например, стерильность, чистота, стабильность и высвобождение лекарственного вещества), которые соответствуют предполагаемому продукту для реализации на рынке.
- Определение критических показателей качества (CQA) можно рассматривать как количественное представление QTPP. В руководстве Q8 (R2) определение CQA гласит: «Физическое, химическое, биологическое или микробиологическое свойство, или характеристика, которые должны соответствовать установленным нормам, диапазону или распределению для обеспечения желаемого качества продукта».
Описание их выведения по результатам работ по разработке приведено в руководстве Q8 (R2).
CQA обычно связаны со следующим:
- Фармацевтическая субстанция
- Вспомогательные вещества
- Полупродукты (внутрипроизводственные материалы)
- Лекарственный препарат
Выявление критических показателей носит итеративный характер и требует как можно более раннего обращения внимания в ходе выполнения работ на то, чтобы вывести из QTPP первоначальный перечень потенциальных CQA для продукта и перейти к изучению предложенного перечня критических характеристик качества продукта, устанавливая критерии приемлемости для тех показателей, которые признаны критическими. Соответствующие CQA могут быть выявлены с помощью итеративного процесса управления рисками для качества (QRM, который является неотъемлемой частью ФСК и экспериментов, оценивающих степень, в которой их изменчивость может повлиять на качество лекарственного препарата).
- Критические характеристики материала (CMA) и параметры процесса (CPP) – определяются как «материал или процесс, вариабельность которых влияет на критический показатель качества, поэтому следует проводить их мониторинг или контролировать для обеспечения желаемого качества лекарственного препарата». Критические показатели качества (CQA) лекарственного препарата и их QTPP учитываются при выборе вспомогательных веществ и параметров процесса.
А именно:
- «Материал» означает сырье, исходные материалы, реактивы, растворители, вспомогательные вещества, промежуточные продукты и т. д., а «Характеристика» означает физическое, химическое, биологическое или микробиологическое свойство, или характеристики данного материала, которые можно измерить. Это может быть морфология частиц, концентрация и тип ингредиентов, соотношение ингредиентов, растворимость в воде, pH и т. д. Характеристика материала имеет критическое значение, когда с большой долей вероятности может повлиять на CQA.
- Критические параметры процесса (CPP) – это еще одна категория факторов риска, связанных с производственными переменными.
Управление рисками качества обычно используется для выявления областей разработки с риском для CQA, однако недостаточно выявить только CPP и CMA. Общая оценка рисков указывает на отдельные операции с риском, а более детальная оценка – выявляет потенциальные факторы, которые могут повлиять на CQA. При этом одной только оценки рисков недостаточно для выявления и контроля CPP. Она может быть основана только на измерении того, как факторы непосредственно влияют на отклик*.
Иными словами, как и для определения критических показателей качества продукта, так и для определения критических параметров процесса необходим научный подход с множеством поставленных экспериментов и статистическим анализом полученных данных.
Разработка стратегии контроля — структурированный подход с участием многопрофильной группы экспертов, связывающих разработку фармацевтической продукции с производственным процессом и инженерно-техническими средствами контроля технологического оборудования. Необходимо тщательно разработать и спланировать мероприятия, чтобы получить продукт, соответствующий установленным показателям качества; поэтому важно получить как можно больше информации о действующих веществах, продукте и производственном процессе, которые подвергаются проверке. Все технологические процессы должны быть разработаны таким образом, чтобы они позволяли стабильно получать продукты, соответствующие критическим показателям качества (CQA), в основном касающимся подлинности, дозировки, качества, чистоты и активности.
По итогу данного процесса компания производитель должна получить эффективную стратегию контроля, отлично понимая, как влияют различные этапы процесса на критические параметры качества продукта, и иметь возможность это доказать. В отличие от подхода «Качество путем контроля» (Quality by Testing), эффективность стратегии заключается и в минимизации контрольных точек в процессе производства, например, если было выявлено, что определенный этап производства не влияет на pH раствора, то нет смысла тратить ресурсы на его контроль после данного этапа.
Масштабирование процесса (Process Scale-Up) – переход от лабораторных / пилотных серий к полномасштабным (коммерческим) сериям. На данном этапе уже должно быть полное понимание процесса и его влияние на качество продукта. В ходе масштабирования проверяются и верифицируются критические параметры процесса. За исключением редких случаев, серии масштабирования являются крайними из немаркетируемых серий. В данных сериях стоит максимально усилить контроль для выявления возможного влияния масштабирования на критические показатели качества продукта (CQAs) и увеличить исследования стабильности препарата для получения максимального количества данных и статистически более обоснованных результатов.
Валидация процесса – данный этап можно по праву назвать самым легким, менее важным и менее трудозатратным в этом списке, ведь доказать, что процесс разработан правильно – легче, чем по факту его разработать. Когда компания использовала при разработке ЛС подход QbD, она досконально изучила свой продукт и процесс его производства. В ходе выполненных экспериментов было получено статистически значимое количество данных по каждому этапу и параметру процесса, а сам процесс имеет высокий индекс воспроизводимости (Process Capability Index).
В данном случае производителю далее следует использовать непрерывную верификацию процесса.
Если первоначальная разработка продукта велась по стандартному подходу, тогда возможно применение гибридного подхода (условно «среднее» между традиционной валидацией и непрерывной верификацией процесса). В данном подходе, основываясь на статистическом анализе данных, которые производитель сумел получить, определяется количество дополнительных серий, которые требуется произвести для увеличения индекса воспроизводимости процесса. Эти дополнительные серии будут считаться «валидационными» и могут стать коммерческими. Количество данных серий напрямую зависит от вариабельности показателей качества продукта и индекса воспроизводимости процесса. Количество валидационных серий продукции на сегодняшний день уже не имеет строгого регулирования в руководящих документах, поэтому общепринятой практикой является определение их числа каждым производителем самостоятельно, применяя риск-ориентированный подход с использованием данных статистического анализа.
Следующие темы заслуживают отдельного внимания:
ПРОСТРАНСТВО ПРОЕКТНЫХ ПАРАМЕТРОВ
В соответствии с ICH Q8 пространство проектных параметров описывает взаимосвязь между «входами» процесса (характеристики сырья и параметры процесса) и критическими показателями качества (CQA).
На практике пространство проектных параметров процесса – это наибольший возможный объем, в пределах которого можно варьировать важные факторы процесса без риска нарушения спецификаций. За пределами этого операционного окна могут возникнуть проблемы с одним или несколькими показателями качества продукта.
Хотя наличие проектного поля не является обязательным, его использование дает ряд несомненных преимуществ:
- Подтверждает высокий уровень понимания процесса
- Демонстрирует и поддерживает гибкость процесса
- Обеспечивает понимание для поддержки предлагаемой стратегии контроля
- Обосновывает меньшее количество испытаний готовой продукции
- Обеспечивает обоснование критериев валидации процесса
- Обеспечивает основу для постоянного совершенствования
Согласно требованиям GMP ЕАЭС, использование данного подхода должно быть зафиксировано в регистрационном досье продукта согласно Рекомендации Коллегии Евразийской экономической комиссии от 26 сентября 2017 г. № 19 «О Руководстве по валидации процесса производства лекарственных препаратов для медицинского применения».
Проектирование экспериментов (Design of Experiments, DoE). Концепция проектирования экспериментов может применяться как на уровне этапа разработки, так и на гораздо более продвинутом этапе в целях улучшения уже существующего процесса. Это означает, что данный инструмент полезен не только на начальных этапах разработки процесса, но также может быть с пользой применен к процессам, уже реализуемым в полноценной GMP-среде.
Концепция DoE полезна в следующем:
- Определение факторов, которые в наибольшей степени влияют на отклик (и)
- Определение тех факторов, которые вместе воздействуют на конкретный отклик – они известны как «взаимодействия»
- Определение оптимальных значений факторов для удовлетворения ряда требуемых откликов.
АНАЛИЗ РИСКОВ
Принципы управления рисками эффективно используются во многих областях фармацевтической отрасли и на разных этапах разработки продуктов, процессов и внедрения в производство и являются ценным компонентом эффективной системы качества.
Деятельность по управлению рисками обычно осуществляется междисциплинарными группами.
Мероприятия управления рисками обычно выполняются на ранних этапах процесса фармацевтической разработки для выявления и ранжирования параметров (например, процесса, оборудования, материалов), которые могут иметь потенциальное влияние на качество продукта. После определения значимых параметров их можно дополнительно изучить для достижения более высокого уровня понимания процесса.
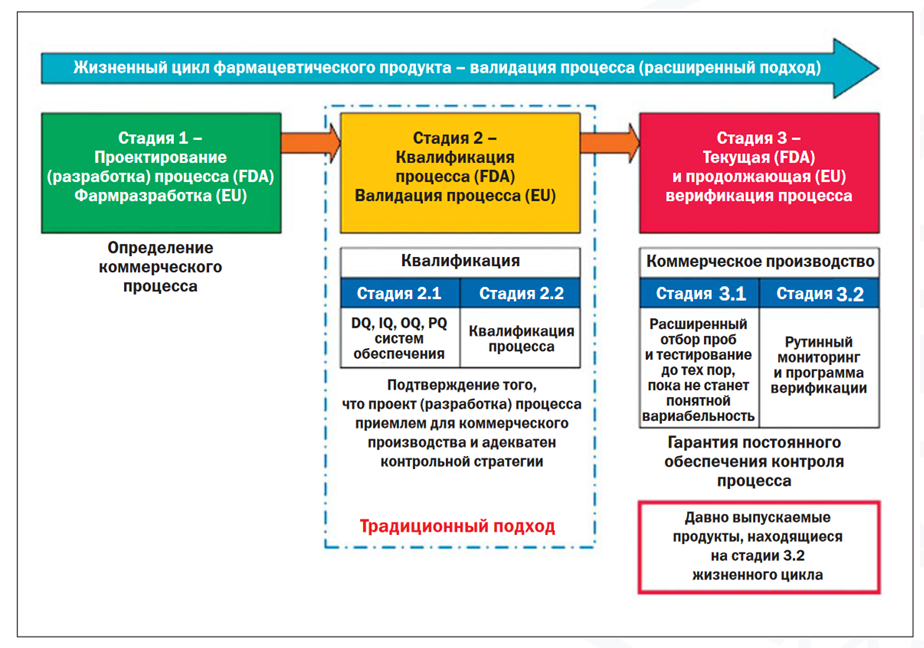
Рис. 2. Стадии жизненного цикла продукта (ISPE GPG: Process validation, стр. 7).
Использование утвержденного подхода к управлению рисками для качества обеспечивает конкретные инструменты для выявления, анализа, оценивания и информирования о рисках на основе знания продукта и развития понимания процесса. Результат управления рисками для качества указывает, где требуется контроль критических параметров и как поддерживать состояние контроля.
При реализации концепта «качество через дизайн» и, соответственно, выработке стратегии контроля ещё на этапе фармразработки, далее речь идёт о верификации этой самой стратегии. В Приложении № 15 GMP ЕАЭС это сформулировано как непрерывная верификация процесса, переходящая в продолжающуюся верификацию процесса в ходе жизненного цикла. Опциональное применение проектного поля (Design Space) имеет ещё одну существенную выгоду – варьирование параметров в рамках определенного проектного поля может не рассматриваться как изменение. Это, в конечном итоге, минимизирует затраты ресурсов как при рассмотрении таких вариаций в рамках процедур управления изменений на предприятии, так и с точки зрения внесения потенциальных изменений в регистрационное досье на препарат – а это уже заметное преимущество. Перерегистрация и внесение изменений в регистрационное досье – процесс, как известно, не быстрый.
В целом жизненный цикл продукта условно разбивается на три стадии (см. рис. 2).
Ввиду того, что валидация процесса перестает быть разовым событием, возрастает важность четко определённых CQA, CPP и верно выработанной стратегии контроля, отражающей особенности процесса. Ведь если это не так, то постоянная фиксация межсерийной вариабельности будет во всех смыслах обременением, влекущим за собой производственные и регуляторные риски, перегруженность лабораторий контроля качества дорогостоящим подтверждением вариабельности по непродуманным стратегиям контроля с риском последующих итераций. Поэтому единственный разумный вариант – прекратить «строить дом с крыши» и с самого начала продумать процесс, так сказать, с «фундамента».
Вместе с тем, руководства ISPE, в частности, рассматривают и случаи, когда продукт уже находится в коммерческой реализации – т. н. Legacy Products. В этом случае продукция начинает свой жизненный цикл с этапа 3.2 (см. рис. 2). Это особенно важный аспект в свете того, что новая версия Приложения № 15 в ЕАЭС вступила в силу только с февраля 2022 года, а до этого момента, безусловно, в коммерческой реализации находилось большое количество зарегистрированных лекарственных препаратов, чья первичная валидация уже была проведена до этого. Для таких препаратов необходимо реализовать продолжающуюся верификацию процесса в ходе жизненного цикла, первично отталкиваясь от данных производства, устанавливая CQA, CPP (если этого не было сделано ранее) и в дальнейшем, при необходимости, внося изменения и/или дополнения в планы отбора проб с целью статистического контроля процесса производства.
Из рациональных предложений в этом плане можно выделить то, что такая деятельность станет частью привычных регулярных обзоров качества продукции с акцентом на обеспечение фиксации событий в режиме реального времени (а не ретроспективно), что позволит при необходимости реализовывать своевременные САРА. Разумеется, в ходе ежегодных обзоров качества исчезнет необходимость дублировать эту деятельность, можно будет просто сослаться на уже построенные тренды и их анализ.
В ходе такой продолжающейся верификации процесса будет возрастать знание в отношении самого процесса, а также будут установлены источники возможной вариабельности. При необходимости, могут быть реализованы изменения, возвращающие процесс как на стадию 2 (повторную валидацию при внесении изменений), так и на стадию 1 (повторную фармразработку).
Для новых лекарственных продуктов настоятельно рекомендуется использовать подход «качество через дизайн», который призван значительно снизить вероятность таких «возвратов» на стадию 1 и стадию 2, что, несомненно, является прямой потерей времени и ресурсов. И пусть фармсообщество не вводит в заблуждение тот факт, что этот подход – «всего лишь» альтернатива «традиционному подходу». Метод проб и ошибок – достаточно дорогостоящий. Именно исключение таких непроизводительных затрат времени и ресурсов является ключевым выходом и ключевой выгодой при следовании подходу «качество через дизайн». Ведь вся суть такого подхода – это статистически значимая «заявка на успех» ещё на этапе фармразработки.
 Орлов Владимир Александрович, Директор Евразийского отделения ISPE: «Одной из ключевых задач ISPE на глобальном уровне является развитие и продвижение современных технических решений и организационных мер, направленных на совершенствование процессов промышленного производства лекарственных средств. Как известно, валидация в целом (как система) и валидация процессов (как ключевой элемент указанной системы) являются базисами построения и поддержания фармацевтической системы качества, позволяющими с определённым уровнем «уверенности» гарантировать качество производимой лекарственной продукции в ходе её жизненного цикла. Однако, безусловно, необходимо поэтапно совершенствовать и обновлять в т. ч. сами подходы и методологию валидации процессов – и здесь важно также придерживаться принципов международной гармонизации в части нормативно-технических требований к производству лекарственных средств. Следуя указанным принципам, при разработке и организации образовательных мероприятий с участием ISPE ЕАЭС особый фокус внимания уделяется тому, чтобы участникам и слушателям были представлены современные, актуальные и востребованные материалы, подготавливаемые ведущими отечественными и зарубежными экспертами».
Орлов Владимир Александрович, Директор Евразийского отделения ISPE: «Одной из ключевых задач ISPE на глобальном уровне является развитие и продвижение современных технических решений и организационных мер, направленных на совершенствование процессов промышленного производства лекарственных средств. Как известно, валидация в целом (как система) и валидация процессов (как ключевой элемент указанной системы) являются базисами построения и поддержания фармацевтической системы качества, позволяющими с определённым уровнем «уверенности» гарантировать качество производимой лекарственной продукции в ходе её жизненного цикла. Однако, безусловно, необходимо поэтапно совершенствовать и обновлять в т. ч. сами подходы и методологию валидации процессов – и здесь важно также придерживаться принципов международной гармонизации в части нормативно-технических требований к производству лекарственных средств. Следуя указанным принципам, при разработке и организации образовательных мероприятий с участием ISPE ЕАЭС особый фокус внимания уделяется тому, чтобы участникам и слушателям были представлены современные, актуальные и востребованные материалы, подготавливаемые ведущими отечественными и зарубежными экспертами».
Пряничникова Оксана Михайловна, Заместитель директора Евразийского отделения ISPE, Директор по развитию PQE CIS: «PQE много лет сотрудничает с ISPE на глобальном уровне, так как верит в роль ISPE в объединении отраслевых знаний и развитии стандартов качества на основе вклада каждого из членов крупнейшей фармацевтической Ассоциации в мире. PQE активно делится своим опытом и активно участвует в международных форумах и конференциях ISPE. Если говорить о локальном опыте, то в качестве примера, эксперт PQE Group из Италии принял участие в 1й Конференции Евразийского отделения ISPE в 2020 году c докладом о целостности данных, а специалисты Евразийского отделения PQE Group Юрий Сандлер и Александр Белинский, не только принимают участие в образовательных программах Академии надлежащих практик, но также являются членами ISPE и активными участниками рабочей группы Евразийского отделения ISPE».
СПРАВОЧНАЯ ИНФОРМАЦИЯ:
Евразийское отделение ISPE – локальное отделение ISPE в Евразийском экономическом союзе, существующее в виде НКО (некоммерческой организации), открытое на базе МАФИ ЕАЭС. Ассоциация создана группой инициативных лиц из фармотрасли, которые объединены идеей продвижения наиболее актуальных и востребованных технологий и идей в области фармацевтического производства, базирующееся на современных аспектах регуляторных требований, технических решений и научных знаний применительно ко всему жизненному циклу лекарственного средства.
PQE Group – международная консалтинговая Группа компаний, офисы представлены в 34 странах мира, Штаб-квартира находится в Италии. Более 20 лет PQE оказывает поддержку мировым производителям лекарственных средств и медицинских изделий в области обеспечения качества.
В странах СНГ PQE работает с 2011 года, за время работы на территории России и ЕАЭС PQE было реализовано более 100 проектов для большинства крупнейших предприятий фарминдустрии. Важным аспектом деятельности PQE является сотрудничество с локальными и международными Ассоциациями и Регуляторами.
 Компания PQE CIS оказывает услуги по поддержке проектов, связанных с внедрением подхода Quality-by-Design, включая построение процессов разработки лекарственных средств от начальных стадий до производства коммерческих серий, а также всех сопутствующих процессов, основанных на научных подходах.
Компания PQE CIS оказывает услуги по поддержке проектов, связанных с внедрением подхода Quality-by-Design, включая построение процессов разработки лекарственных средств от начальных стадий до производства коммерческих серий, а также всех сопутствующих процессов, основанных на научных подходах.
Чтобы узнать больше, свяжитесь с представителем PQE CIS в России по электронной почте ru.info@pqegroup.com
ООО «ПИКЬЮИ СИАЙЭС»
127015, г.Москва ул. Новодмитровская д.2, к.2 БЦ Савеловский Сити, башня Davis.
Тел.: +7 (495) 133 — 98 — 36






 Контакты:
Контакты:





 ЧИСТЫЕ ПОМЕЩЕНИЯ
ЧИСТЫЕ ПОМЕЩЕНИЯ Впрочем, даже с учётом авторской оговорки в отношении величин Fh и Fd, укажу, что расчёт и того, и другого значения не критичен. Для этого копнём чуть глубже и проанализируем расчётные формулы обеих величин, Fh и Fd [4]:
Впрочем, даже с учётом авторской оговорки в отношении величин Fh и Fd, укажу, что расчёт и того, и другого значения не критичен. Для этого копнём чуть глубже и проанализируем расчётные формулы обеих величин, Fh и Fd [4]:













 Сфера деятельности IBC Clean-systems:
Сфера деятельности IBC Clean-systems: Все компоненты, применяемые в панели PSW делают ее полностью герметичной, исключающей возможность контаминации и применимой в чистых помещениях.
Все компоненты, применяемые в панели PSW делают ее полностью герметичной, исключающей возможность контаминации и применимой в чистых помещениях.